BNPP
Inhibitor of carboxylesterases (and also AADAC), substrate of metallo-beta-lactamase. hCE1b IC50 69.3 +/- 9.4
General
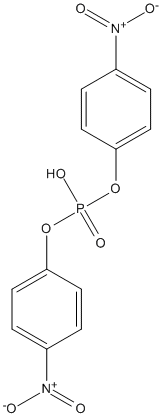
Type : Organophosphate,pNP
Chemical_Nomenclature : Bis(4-nitrophenyl)phosphate
Canonical SMILES : C1=CC(=CC=C1[N+](=O)[O-])OP(=O)(O)OC2=CC=C(C=C2)[N+](=O)[O-]
InChI : InChI=1S\/C12H9N2O8P\/c15-13(16)9-1-5-11(6-2-9)21-23(19,20)22-12-7-3-10(4-8-12)14(17)18\/h1-8H,(H,19,20)
InChIKey : MHSVUSZEHNVFKW-UHFFFAOYSA-N
Other name(s) : Bis(4-nitrophenyl)phosphate,Bis(4-nitrophenyl) phosphate,Bis(4-nitrophenyl) hydrogen phosphate,Bis(p-nitrophenyl)phosphate,bis-(p-nitrophenyl)phosphoric acid
MW : 340.18
Formula : C12H9N2O8P
CAS_number : 645-15-8
PubChem : 255
UniChem : MHSVUSZEHNVFKW-UHFFFAOYSA-N
IUPHAR :
Wikipedia : Bis_(p-nitrophenyl)phosphate
Target
Families : BNPP ligand of proteins in family: Carb_B_Chordata || Arylacetamide_deacetylase
Stucture :
Protein : human-CES2
References (11)
| Title : Rationally Engineered hCES2A Near-Infrared Fluorogenic Substrate for Functional Imaging and High-Throughput Inhibitor Screening - Fan_2023_Anal.Chem_95_15665 |
| Author(s) : Fan Y , Zhang T , Song Y , Sang Z , Zeng H , Liu P , Wang P , Ge G |
| Ref : Analytical Chemistry , 95 :15665-15672 , 2023 |
| Abstract : Fan_2023_Anal.Chem_95_15665 |
| ESTHER : Fan_2023_Anal.Chem_95_15665 |
| PubMedSearch : Fan_2023_Anal.Chem_95_15665 |
| PubMedID: 37782032 |
| Gene_locus related to this paper: human-CES2 |
| Title : Deciphering the species differences in CES1A-mediated hydrolytic metabolism by using a bioluminescence substrate - Jin_2022_Chem.Biol.Interact_368_110197 |
| Author(s) : Jin Q , Li Z , Zhang MJ , Liu WC , Zou LW , Sui H , Wang DD , Tang QF , Ge GB |
| Ref : Chemico-Biological Interactions , 368 :110197 , 2022 |
| Abstract : Jin_2022_Chem.Biol.Interact_368_110197 |
| ESTHER : Jin_2022_Chem.Biol.Interact_368_110197 |
| PubMedSearch : Jin_2022_Chem.Biol.Interact_368_110197 |
| PubMedID: 36174736 |
| Title : Optimal pH 8.5 to 9 for the Hydrolysis of Vixotrigine and Other Basic Substrates of Carboxylesterase-1 in Human Liver Microsomes - Johnson_2022_Xenobiotica_52_105 |
| Author(s) : Johnson JL , Huang J , Rooney M , Gu C |
| Ref : Xenobiotica , 52 :105 , 2021 |
| Abstract : Johnson_2022_Xenobiotica_52_105 |
| ESTHER : Johnson_2022_Xenobiotica_52_105 |
| PubMedSearch : Johnson_2022_Xenobiotica_52_105 |
| PubMedID: 34904522 |
| Title : Inhibition of Human Liver Carboxylesterase (hCE1) by Organophosphate Ester Flame Retardants and Plasticizers: Implications for Pharmacotherapy - Phillips_2019_Toxicol.Sci_171_396 |
| Author(s) : Phillips AL , Stapleton HM |
| Ref : Toxicol Sci , 171 :396 , 2019 |
| Abstract : Phillips_2019_Toxicol.Sci_171_396 |
| ESTHER : Phillips_2019_Toxicol.Sci_171_396 |
| PubMedSearch : Phillips_2019_Toxicol.Sci_171_396 |
| PubMedID: 31268531 |
| Title : Inhibition of human carboxylesterases hCE1 and hiCE by cholinesterase inhibitors - Tsurkan_2013_Chem.Biol.Interact_203_226 |
| Author(s) : Tsurkan LG , Hatfield MJ , Edwards CC , Hyatt JL , Potter PM |
| Ref : Chemico-Biological Interactions , 203 :226 , 2013 |
| Abstract : Tsurkan_2013_Chem.Biol.Interact_203_226 |
| ESTHER : Tsurkan_2013_Chem.Biol.Interact_203_226 |
| PubMedSearch : Tsurkan_2013_Chem.Biol.Interact_203_226 |
| PubMedID: 23123248 |
| Title : Comparative study of the hydrolytic metabolism of methyl-, ethyl-, propyl-, butyl-, heptyl- and dodecylparaben by microsomes of various rat and human tissues - Ozaki_2013_Xenobiotica_43_1064 |
| Author(s) : Ozaki H , Sugihara K , Watanabe Y , Fujino C , Uramaru N , Sone T , Ohta S , Kitamura S |
| Ref : Xenobiotica , 43 :1064 , 2013 |
| Abstract : Ozaki_2013_Xenobiotica_43_1064 |
| ESTHER : Ozaki_2013_Xenobiotica_43_1064 |
| PubMedSearch : Ozaki_2013_Xenobiotica_43_1064 |
| PubMedID: 23742084 |
| Title : Utility of the carboxylesterase inhibitor bis-para-nitrophenylphosphate (BNPP) in the plasma unbound fraction determination for a hydrolytically unstable amide derivative and agonist of the TGR5 receptor - Eng_2010_Xenobiotica_40_369 |
| Author(s) : Eng H , Niosi M , McDonald TS , Wolford A , Chen Y , Simila ST , Bauman JN , Warmus J , Kalgutkar AS |
| Ref : Xenobiotica , 40 :369 , 2010 |
| Abstract : Eng_2010_Xenobiotica_40_369 |
| ESTHER : Eng_2010_Xenobiotica_40_369 |
| PubMedSearch : Eng_2010_Xenobiotica_40_369 |
| PubMedID: 20297923 |
| Title : Human arylacetamide deacetylase is a principal enzyme in flutamide hydrolysis - Watanabe_2009_Drug.Metab.Dispos_37_1513 |
| Author(s) : Watanabe A , Fukami T , Nakajima M , Takamiya M , Aoki Y , Yokoi T |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 37 :1513 , 2009 |
| Abstract : Watanabe_2009_Drug.Metab.Dispos_37_1513 |
| ESTHER : Watanabe_2009_Drug.Metab.Dispos_37_1513 |
| PubMedSearch : Watanabe_2009_Drug.Metab.Dispos_37_1513 |
| PubMedID: 19339378 |
| Gene_locus related to this paper: human-AADAC |
| Title : Involvement of human blood arylesterases and liver microsomal carboxylesterases in nafamostat hydrolysis - Yamaori_2006_Drug.Metab.Pharmacokinet_21_147 |
| Author(s) : Yamaori S , Fujiyama N , Kushihara M , Funahashi T , Kimura T , Yamamoto I , Sone T , Isobe M , Ohshima T , Matsumura K , Oda M , Watanabe K |
| Ref : Drug Metab Pharmacokinet , 21 :147 , 2006 |
| Abstract : Yamaori_2006_Drug.Metab.Pharmacokinet_21_147 |
| ESTHER : Yamaori_2006_Drug.Metab.Pharmacokinet_21_147 |
| PubMedSearch : Yamaori_2006_Drug.Metab.Pharmacokinet_21_147 |
| PubMedID: 16702735 |
| Title : Detection of a new N-oxidized metabolite of flutamide, N-[4-nitro-3-(trifluoromethyl)phenyl]hydroxylamine, in human liver microsomes and urine of prostate cancer patients - Goda_2006_Drug.Metab.Dispos_34_828 |
| Author(s) : Goda R , Nagai D , Akiyama Y , Nishikawa K , Ikemoto I , Aizawa Y , Nagata K , Yamazoe Y |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 34 :828 , 2006 |
| Abstract : Goda_2006_Drug.Metab.Dispos_34_828 |
| ESTHER : Goda_2006_Drug.Metab.Dispos_34_828 |
| PubMedSearch : Goda_2006_Drug.Metab.Dispos_34_828 |
| PubMedID: 16507648 |