Carmegliptin
Carmegliptin is a potent, long-acting, selective, orally bioavailable, pyrrolidinone-based inhibitor of dipeptidyl peptidase 4 (DPP-4), with hypoglycemic activity. Carmegliptin shows extensive tissue distribution and is excreted unchanged in the urine and bile.
General
Type : Drug,Pyrrolidine,Gliptin,Quinolizine
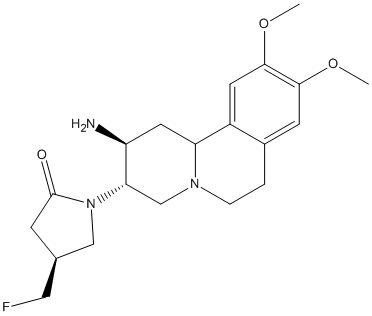
Chemical_Nomenclature : (4S)-1-[(2S,3S,11bS)-2-amino-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-benzo[a]quinolizin-3-yl]-4-(fluoromethyl)pyrrolidin-2-one
Canonical SMILES : COC1=C(C=C2C3CC(C(CN3CCC2=C1)N4CC(CC4=O)CF)N)OC
InChI : InChI=1S\/C20H28FN3O3\/c1-26-18-6-13-3-4-23-11-17(24-10-12(9-21)5-20(24)25)15(22)8-16(23)14(13)7-19(18)27-2\/h6-7,12,15-17H,3-5,8-11,22H2,1-2H3\/t12-,15+,16+,17+\/m1\/s1
InChIKey : GUYMHFIHHOEFOA-ZCPGHIKRSA-N
Other name(s) : B1Q,UNII-9Z723VGH7J,R-1579,CHEMBL591118
MW : 377.46
Formula : C20H28FN3O3
CAS_number : 813452-18-5
PubChem : 11417567
UniChem : GUYMHFIHHOEFOA-ZCPGHIKRSA-N
IUPHAR :
Wikipedia :
Target
Families : Carmegliptin ligand of proteins in family: DPP4N_Peptidase_S9
Stucture : 3KWF human DPP-IV with carmegliptin (S)-1-((2S,3S,11bS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one
Protein : human-DPP4
References (5)
| Title : Retrospective and Prospective Human Intravenous and Oral Pharmacokinetic Projection of Dipeptidyl peptidase-IV Inhibitors Using Simple Allometric Principles - Case Studies of ABT-279, ABT-341, Alogliptin, Carmegliptin, Sitagliptin and Vildagliptin - Gilibili_2015_J.Pharm.Pharm.Sci_18_434 |
| Author(s) : Gilibili RR , Bhamidipati RK , Mullangi R , Srinivas NR |
| Ref : J Pharm Pharm Sci , 18 :434 , 2015 |
| Abstract : Gilibili_2015_J.Pharm.Pharm.Sci_18_434 |
| ESTHER : Gilibili_2015_J.Pharm.Pharm.Sci_18_434 |
| PubMedSearch : Gilibili_2015_J.Pharm.Pharm.Sci_18_434 |
| PubMedID: 26517136 |
| Title : Interaction potential of Carmegliptin with P-glycoprotein (Pgp) transporter in healthy volunteers - Kuhlmann_2014_J.Drug.Assess_3_28 |
| Author(s) : Kuhlmann O , Carlile D , Noe J , Bentley D |
| Ref : J Drug Assess , 3 :28 , 2014 |
| Abstract : Kuhlmann_2014_J.Drug.Assess_3_28 |
| ESTHER : Kuhlmann_2014_J.Drug.Assess_3_28 |
| PubMedSearch : Kuhlmann_2014_J.Drug.Assess_3_28 |
| PubMedID: 27536451 |
| Title : Ligand-based pharmacophore detection, screening of potential gliptins and docking studies to get effective antidiabetic agents - Agrawal_2012_Comb.Chem.High.Throughput.Screen_15_849 |
| Author(s) : Agrawal R , Jain P , Dikshit SN |
| Ref : Comb Chem High Throughput Screen , 15 :849 , 2012 |
| Abstract : Agrawal_2012_Comb.Chem.High.Throughput.Screen_15_849 |
| ESTHER : Agrawal_2012_Comb.Chem.High.Throughput.Screen_15_849 |
| PubMedSearch : Agrawal_2012_Comb.Chem.High.Throughput.Screen_15_849 |
| PubMedID: 23140189 |
| Title : Pharmacokinetics and metabolism of the dipeptidyl peptidase IV inhibitor carmegliptin in rats, dogs, and monkeys - Kuhlmann_2010_Xenobiotica_40_840 |
| Author(s) : Kuhlmann O , Paehler A , Weick I , Funk C , Pantze M , Zell M , Timm U |
| Ref : Xenobiotica , 40 :840 , 2010 |
| Abstract : Kuhlmann_2010_Xenobiotica_40_840 |
| ESTHER : Kuhlmann_2010_Xenobiotica_40_840 |
| PubMedSearch : Kuhlmann_2010_Xenobiotica_40_840 |
| PubMedID: 20868265 |
| Title : Discovery of carmegliptin: a potent and long-acting dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes - Mattei_2010_Bioorg.Med.Chem.Lett_20_1109 |
| Author(s) : Mattei P , Boehringer M , Di Giorgio P , Fischer H , Hennig M , Huwyler J , Kocer B , Kuhn B , Loeffler BM , Macdonald A , Narquizian R , Rauber E , Sebokova E , Sprecher U |
| Ref : Bioorganic & Medicinal Chemistry Lett , 20 :1109 , 2010 |
| Abstract : Mattei_2010_Bioorg.Med.Chem.Lett_20_1109 |
| ESTHER : Mattei_2010_Bioorg.Med.Chem.Lett_20_1109 |
| PubMedSearch : Mattei_2010_Bioorg.Med.Chem.Lett_20_1109 |
| PubMedID: 20031405 |
| Gene_locus related to this paper: human-DPP4 |