DFP
Inhibitor of cholinesterases and many serine hydrolases (not only alpha/beta hydrolases but also trypsin chymotrypsin etc.). DFP is a parasympathomimetic drug used in ophthalmology as a miotic agent in treatment of chronic glaucoma, as a miotic in veterinary medicine, and as an experimental agent in neuroscience because of its acetylcholinesterase inhibitory properties and ability to induce delayed peripheral neuropathy. DFP can phosphorylate other residues than active serine (tyrosine, lysine) see pdb 1TRN and Grigoryan et al. BChE appears to be about 100-200 times as sensitive as AChE to DFP and DFP was used to distinguish AChE and BChE before BW284C51 and Iso-OMPA were discovered. DFP is the oxygen analogue of Mipafox.(Structures of not alpha/beta hydrolases inhibited by DFP include: 4V08, 4H1D, 3VTA, 3TT7, 3TXT, 3MH5, 3MH6, 1RS0, 1CI9, 1DUI, 1AT3, 1SUE, 1DFP, 2TGD, 4PKA, 3LH3, 3GCN, 3GCO, 3GDS, 3GDU, 3GDV, 2RCE, 2Z2X, 1PPZ, 1GNV, 1A2Q, 5PTP)
General
Type : Organophosphate
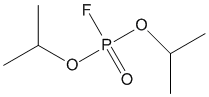
Chemical_Nomenclature : Diisopropylphosphorofluoridate
Canonical SMILES : CC(C)OP(=O)(OC(C)C)F
InChI : InChI=1S\/C6H14FO3P\/c1-5(2)9-11(7,8)10-6(3)4\/h5-6H,1-4H3
InChIKey : MUCZHBLJLSDCSD-UHFFFAOYSA-N
Other name(s) : Diisopropylphosphorofluoridate,Dyflos,Diflupyl,Floropyl,isofluorophate,Fluostigmine,Isoflurophate,Difluorophate,Diflurphate,Dyphlos,isofluorphate,Neoglaucit,PF-3,PF3,T-1703,TL 466
MW : 184.1
Formula : C6H14FO3P
CAS_number : 55-91-4
PubChem : 5936
UniChem : MUCZHBLJLSDCSD-UHFFFAOYSA-N
IUPHAR :
Wikipedia : Diisopropyl_fluorophosphate
Target
Families : DFP ligand of proteins in family: PAF-Acetylhydrolase || A85-IroE-IroD-Fes-Yiel || DPP4N_Peptidase_S9 || Cutinase || ACHE || BCHE || Peptidase_S15 || Lactobacillus_peptidase || Cocaine_esterase || Prolylcarboxypeptidase || PolyAspartate-hydrolase || ACPH_Peptidase_S9 || 6_AlphaBeta_hydrolase || Carb_B_Chordata || Carb_B_Arthropoda || Arylacetamide_deacetylase || Carboxypeptidase_S10 || Lipase_3
Stucture :
References (56)
| Title : Structural and Kinetic Evidence of Aging after Organophosphate Inhibition of Human Cathepsin A - Bouknight_2020_Biochem.Pharmacol__113980 |
| Author(s) : Bouknight KD , Jurkouich KM , Compton JR , Khavrutskii IV , Guelta MA , Harvey SP , Legler PM |
| Ref : Biochemical Pharmacology , :113980 , 2020 |
| Abstract : Bouknight_2020_Biochem.Pharmacol__113980 |
| ESTHER : Bouknight_2020_Biochem.Pharmacol__113980 |
| PubMedSearch : Bouknight_2020_Biochem.Pharmacol__113980 |
| PubMedID: 32305437 |
| Gene_locus related to this paper: human-CTSA |
| Title : Discovery of New Classes of Compounds that Reactivate Acetylcholinesterase Inhibited by Organophosphates - Katz_2015_Chembiochem_16_2205 |
| Author(s) : Katz FS , Pecic S , Tran TH , Trakht I , Schneider L , Zhu Z , Ton-That L , Luzac M , Zlatanic V , Damera S , MacDonald J , Landry DW , Tong L , Stojanovic MN |
| Ref : Chembiochem , 16 :2205 , 2015 |
| Abstract : Katz_2015_Chembiochem_16_2205 |
| ESTHER : Katz_2015_Chembiochem_16_2205 |
| PubMedSearch : Katz_2015_Chembiochem_16_2205 |
| PubMedID: 26350723 |
| Gene_locus related to this paper: mouse-ACHE |
| Title : Structural characterization of enterobactin hydrolase IroE - Larsen_2006_Biochemistry_45_10184 |
| Author(s) : Larsen NA , Lin H , Wei R , Fischbach MA , Walsh CT |
| Ref : Biochemistry , 45 :10184 , 2006 |
| Abstract : Larsen_2006_Biochemistry_45_10184 |
| ESTHER : Larsen_2006_Biochemistry_45_10184 |
| PubMedSearch : Larsen_2006_Biochemistry_45_10184 |
| PubMedID: 16922493 |
| Gene_locus related to this paper: ecoli-IROE |
| Title : Role of water in aging of human butyrylcholinesterase inhibited by echothiophate: the crystal structure suggests two alternative mechanisms of aging - Nachon_2005_Biochemistry_44_1154 |
| Author(s) : Nachon F , Asojo OA , Borgstahl GE , Masson P , Lockridge O |
| Ref : Biochemistry , 44 :1154 , 2005 |
| Abstract : Nachon_2005_Biochemistry_44_1154 |
| ESTHER : Nachon_2005_Biochemistry_44_1154 |
| PubMedSearch : Nachon_2005_Biochemistry_44_1154 |
| PubMedID: 15667209 |
| Gene_locus related to this paper: human-BCHE |
| Title : Tyrosine 547 constitutes an essential part of the catalytic mechanism of dipeptidyl peptidase IV - Bjelke_2004_J.Biol.Chem_279_34691 |
| Author(s) : Bjelke JR , Christensen J , Branner S , Wagtmann N , Olsen C , Kanstrup AB , Rasmussen HB |
| Ref : Journal of Biological Chemistry , 279 :34691 , 2004 |
| Abstract : Bjelke_2004_J.Biol.Chem_279_34691 |
| ESTHER : Bjelke_2004_J.Biol.Chem_279_34691 |
| PubMedSearch : Bjelke_2004_J.Biol.Chem_279_34691 |
| PubMedID: 15175333 |
| Gene_locus related to this paper: human-DPP4 |
| Title : Effects of mutations of active site residues and amino acids interacting with the Omega loop on substrate activation of butyrylcholinesterase - Masson_2001_Biochim.Biophys.Acta_1544_166 |
| Author(s) : Masson P , Xie W , Froment MT , Lockridge O |
| Ref : Biochimica & Biophysica Acta , 1544 :166 , 2001 |
| Abstract : Masson_2001_Biochim.Biophys.Acta_1544_166 |
| ESTHER : Masson_2001_Biochim.Biophys.Acta_1544_166 |
| PubMedSearch : Masson_2001_Biochim.Biophys.Acta_1544_166 |
| PubMedID: 11341926 |
| Gene_locus related to this paper: human-BCHE |
| Title : Mutagenesis of organophosphorus hydrolase to enhance hydrolysis of the nerve agent VX - Gopal_2000_Biochem.Biophys.Res.Commun_279_516 |
| Author(s) : Gopal S , Rastogi V , Ashman W , Mulbry W |
| Ref : Biochemical & Biophysical Research Communications , 279 :516 , 2000 |
| Abstract : Gopal_2000_Biochem.Biophys.Res.Commun_279_516 |
| ESTHER : Gopal_2000_Biochem.Biophys.Res.Commun_279_516 |
| PubMedSearch : Gopal_2000_Biochem.Biophys.Res.Commun_279_516 |
| PubMedID: 11118318 |
| Title : Evidence for homeostatic adjustments of rat somatosensory cortical neurons to changes in extracellular acetylcholine concentrations produced by iontophoretic administration of acetylcholine and by systemic diisopropylfluorophosphate treatment - Testylier_1999_Neurosci_91_843 |
| Author(s) : Testylier G , Maalouf M , Butt AE , Miasnikov AA , Dykes RW |
| Ref : Neuroscience , 91 :843 , 1999 |
| Abstract : Testylier_1999_Neurosci_91_843 |
| ESTHER : Testylier_1999_Neurosci_91_843 |
| PubMedSearch : Testylier_1999_Neurosci_91_843 |
| PubMedID: 10391467 |
| Title : Crystal structures of aged phosphonylated acetylcholinesterase: nerve agent reaction products at the atomic level - Millard_1999_Biochemistry_38_7032 |
| Author(s) : Millard CB , Kryger G , Ordentlich A , Greenblatt HM , Harel M , Raves ML , Segall Y , Barak D , Shafferman A , Silman I , Sussman JL |
| Ref : Biochemistry , 38 :7032 , 1999 |
| Abstract : Millard_1999_Biochemistry_38_7032 |
| ESTHER : Millard_1999_Biochemistry_38_7032 |
| PubMedSearch : Millard_1999_Biochemistry_38_7032 |
| PubMedID: 10353814 |
| Gene_locus related to this paper: torca-ACHE |
| Title : Identification of serine esterases in tissue homogenates - Keshavarz-Shokri_1999_Anal.Biochem_267_406 |
| Author(s) : Keshavarz-Shokri A , Suntornwat O , Kitos PA |
| Ref : Analytical Biochemistry , 267 :406 , 1999 |
| Abstract : Keshavarz-Shokri_1999_Anal.Biochem_267_406 |
| ESTHER : Keshavarz-Shokri_1999_Anal.Biochem_267_406 |
| PubMedSearch : Keshavarz-Shokri_1999_Anal.Biochem_267_406 |
| PubMedID: 10036148 |
| Title : Modification of the rate of aging of diisopropylfluorophosphate- inhibited neuropathy target esterase of hen brain - Jokanovic_1998_Toxicol.Lett_95_93 |
| Author(s) : Jokanovic M , Stepanovic RM , Maksimovic M , Kosanovic M , Stojiljkovic MP |
| Ref : Toxicol Lett , 95 :93 , 1998 |
| Abstract : Jokanovic_1998_Toxicol.Lett_95_93 |
| ESTHER : Jokanovic_1998_Toxicol.Lett_95_93 |
| PubMedSearch : Jokanovic_1998_Toxicol.Lett_95_93 |
| PubMedID: 9635412 |
| Title : Identification of active site residues in a ferulic acid esterase (FAE-III) from Aspergillus niger - |
| Author(s) : Aliwan FO , Williamson G |
| Ref : Biochemical Society Transactions , 26 :S164 , 1998 |
| PubMedID: 9649839 |
| Gene_locus related to this paper: aspni-FAEA |
| Title : Inhibition of carboxylesterases in SH-SY5Y human and NB41A3 mouse neuroblastoma cells by organophosphorus esters - Ehrich_1998_J.Toxicol.Environ.Health_53_385 |
| Author(s) : Ehrich M , Correll L |
| Ref : J Toxicol Environ Health , 53 :385 , 1998 |
| Abstract : Ehrich_1998_J.Toxicol.Environ.Health_53_385 |
| ESTHER : Ehrich_1998_J.Toxicol.Environ.Health_53_385 |
| PubMedSearch : Ehrich_1998_J.Toxicol.Environ.Health_53_385 |
| PubMedID: 9515941 |
| Title : Augmented hydrolysis of diisopropyl fluorophosphate in engineered mutants of phosphotriesterase - Watkins_1997_J.Biol.Chem_272_25596 |
| Author(s) : Watkins LM , Mahoney HJ , McCulloch JK , Raushel FM |
| Ref : Journal of Biological Chemistry , 272 :25596 , 1997 |
| Abstract : Watkins_1997_J.Biol.Chem_272_25596 |
| ESTHER : Watkins_1997_J.Biol.Chem_272_25596 |
| PubMedSearch : Watkins_1997_J.Biol.Chem_272_25596 |
| PubMedID: 9325279 |
| Title : Importance of aspartate-70 in organophosphate inhibition, oxime re-activation and aging of human butyrylcholinesterase - Masson_1997_Biochem.J_325_53 |
| Author(s) : Masson P , Froment MT , Bartels CF , Lockridge O |
| Ref : Biochemical Journal , 325 :53 , 1997 |
| Abstract : Masson_1997_Biochem.J_325_53 |
| ESTHER : Masson_1997_Biochem.J_325_53 |
| PubMedSearch : Masson_1997_Biochem.J_325_53 |
| PubMedID: 9224629 |
| Title : Down-regulation of cardiac muscarinic receptors induced by di- isopropylfluorophosphate - Valette_1997_J.Nucl.Med_38_1430 |
| Author(s) : Valette H , Syrota A , Fuseau C |
| Ref : J Nucl Med , 38 :1430 , 1997 |
| Abstract : Valette_1997_J.Nucl.Med_38_1430 |
| ESTHER : Valette_1997_J.Nucl.Med_38_1430 |
| PubMedSearch : Valette_1997_J.Nucl.Med_38_1430 |
| PubMedID: 9293803 |
| Title : Characterization of monoclonal antibodies that inhibit the catalytic activity of acetylcholinesterases - Gentry_1995_J.Neurochem_64_842 |
| Author(s) : Gentry MK , Moorad DR , Hur RS , Saxena A , Ashani Y , Doctor BP |
| Ref : Journal of Neurochemistry , 64 :842 , 1995 |
| Abstract : Gentry_1995_J.Neurochem_64_842 |
| ESTHER : Gentry_1995_J.Neurochem_64_842 |
| PubMedSearch : Gentry_1995_J.Neurochem_64_842 |
| PubMedID: 7830078 |
| Title : Comparison of the relative inhibition of acetylcholinesterase and neuropathy target esterase in rats and hens given cholinesterase inhibitors - Ehrich_1995_Fundam.Appl.Toxicol_24_94 |
| Author(s) : Ehrich M , Jortner BS , Padilla S |
| Ref : Fundamental & Applied Toxicology , 24 :94 , 1995 |
| Abstract : Ehrich_1995_Fundam.Appl.Toxicol_24_94 |
| ESTHER : Ehrich_1995_Fundam.Appl.Toxicol_24_94 |
| PubMedSearch : Ehrich_1995_Fundam.Appl.Toxicol_24_94 |
| PubMedID: 7713347 |
| Title : Strain differences in the laboratory rat: impact on the autonomic, behavioral, and biochemical response to cholinesterase inhibition - Gordon_1995_J.Toxicol.Env.Health_45_59 |
| Author(s) : Gordon CJ , Watkinson WP |
| Ref : Journal of Toxicology & Environmental Health , 45 :59 , 1995 |
| Abstract : Gordon_1995_J.Toxicol.Env.Health_45_59 |
| ESTHER : Gordon_1995_J.Toxicol.Env.Health_45_59 |
| PubMedSearch : Gordon_1995_J.Toxicol.Env.Health_45_59 |
| PubMedID: 7752289 |
| Title : Raman spectroscopic study of conjugates of butyrylcholinesterase with organophosphates - Aslanian_1995_Biochim.Biophys.Acta_1249_37 |
| Author(s) : Aslanian D , Grof P , Renault F , Masson P |
| Ref : Biochimica & Biophysica Acta , 1249 :37 , 1995 |
| Abstract : Aslanian_1995_Biochim.Biophys.Acta_1249_37 |
| ESTHER : Aslanian_1995_Biochim.Biophys.Acta_1249_37 |
| PubMedSearch : Aslanian_1995_Biochim.Biophys.Acta_1249_37 |
| PubMedID: 7766682 |
| Title : Allosteric modulation of acetylcholinesterase activity by peripheral ligands involves a conformational transition of the anionic subsite - Barak_1995_Biochemistry_34_15444 |
| Author(s) : Barak D , Ordentlich A , Bromberg A , Kronman C , Marcus D , Lazar A , Ariel N , Velan B , Shafferman A |
| Ref : Biochemistry , 34 :15444 , 1995 |
| Abstract : Barak_1995_Biochemistry_34_15444 |
| ESTHER : Barak_1995_Biochemistry_34_15444 |
| PubMedSearch : Barak_1995_Biochemistry_34_15444 |
| PubMedID: 7492545 |
| Title : Site-directed mutagenesis of active site residues reveals plasticity of human butyrylcholinesterase in substrate and inhibitor interactions - Gnatt_1994_J.Neurochem_62_749 |
| Author(s) : Gnatt A , Loewenstein Y , Yaron A , Schwarz M , Soreq H |
| Ref : Journal of Neurochemistry , 62 :749 , 1994 |
| Abstract : Gnatt_1994_J.Neurochem_62_749 |
| ESTHER : Gnatt_1994_J.Neurochem_62_749 |
| PubMedSearch : Gnatt_1994_J.Neurochem_62_749 |
| PubMedID: 8294937 |
| Title : The timing of channel opening during miniature endplate currents at the frog and mouse neuromuscular junctions: effects of fasciculin-2, other anti-cholinesterases and vesamicol - Van der Kloot_1994_Pflugers.Arch_428_114 |
| Author(s) : Van der Kloot W , Balezina OP , Molgo J , Naves LA |
| Ref : Pflugers Arch , 428 :114 , 1994 |
| Abstract : Van der Kloot_1994_Pflugers.Arch_428_114 |
| ESTHER : Van der Kloot_1994_Pflugers.Arch_428_114 |
| PubMedSearch : Van der Kloot_1994_Pflugers.Arch_428_114 |
| PubMedID: 7971167 |
| Title : [A kinetic study of the blood serum cholinesterase activity in the freshwater fish Abramis ballerus] - Zhukovskii_1994_Zh.Evol.Biokhim.Fiziol_30_177 |
| Author(s) : Zhukovskii Iu G , Kutsenko SA , Kuznetsova LP , Sochilina EE , Dmitrieva EN , Fartseiger NL , Tonkopii VD , Korkishko NN , Kozlovskaia VI , Fel'd VE |
| Ref : Zh Evol Biokhim Fiziol , 30 :177 , 1994 |
| Abstract : Zhukovskii_1994_Zh.Evol.Biokhim.Fiziol_30_177 |
| ESTHER : Zhukovskii_1994_Zh.Evol.Biokhim.Fiziol_30_177 |
| PubMedSearch : Zhukovskii_1994_Zh.Evol.Biokhim.Fiziol_30_177 |
| PubMedID: 7817653 |
| Title : Choline derivatives and sodium fluoride protect acetylcholinesterase against irreversible inhibition and aging by DFP and paraoxon - Dehlawi_1994_J.Biochem.Toxicol_9_261 |
| Author(s) : Dehlawi MS , Eldefrawi AT , Eldefrawi ME , Anis NA , Valdes JJ |
| Ref : Journal of Biochemical Toxicology , 9 :261 , 1994 |
| Abstract : Dehlawi_1994_J.Biochem.Toxicol_9_261 |
| ESTHER : Dehlawi_1994_J.Biochem.Toxicol_9_261 |
| PubMedSearch : Dehlawi_1994_J.Biochem.Toxicol_9_261 |
| PubMedID: 7853361 |
| Title : Phosphotriesterase--a promising candidate for use in detoxification of organophosphates - Tuovinen_1994_Fundam.Appl.Toxicol_23_578 |
| Author(s) : Tuovinen K , Kaliste-Korhonen E , Raushel FM , Hanninen O |
| Ref : Fundamental & Applied Toxicology , 23 :578 , 1994 |
| Abstract : Tuovinen_1994_Fundam.Appl.Toxicol_23_578 |
| ESTHER : Tuovinen_1994_Fundam.Appl.Toxicol_23_578 |
| PubMedSearch : Tuovinen_1994_Fundam.Appl.Toxicol_23_578 |
| PubMedID: 7867909 |
| Title : Physiologically based pharmacokinetic model for the inhibition of acetylcholinesterase by organophosphate esters - Gearhart_1994_Environ.Health.Perspect_11_51 |
| Author(s) : Gearhart JM , Jepson GW , Clewell HJ , Andersen ME , Conolly RB |
| Ref : Environmental Health Perspectives , 11 :51 , 1994 |
| Abstract : Gearhart_1994_Environ.Health.Perspect_11_51 |
| ESTHER : Gearhart_1994_Environ.Health.Perspect_11_51 |
| PubMedSearch : Gearhart_1994_Environ.Health.Perspect_11_51 |
| PubMedID: 7737042 |
| Title : The molecular forms of acetylcholinesterase from Necator americanus (Nematoda), a hookworm parasite of the human intestine - Pritchard_1994_Eur.J.Biochem_219_317 |
| Author(s) : Pritchard DI , Brown A , Toutant JP |
| Ref : European Journal of Biochemistry , 219 :317 , 1994 |
| Abstract : Pritchard_1994_Eur.J.Biochem_219_317 |
| ESTHER : Pritchard_1994_Eur.J.Biochem_219_317 |
| PubMedSearch : Pritchard_1994_Eur.J.Biochem_219_317 |
| PubMedID: 8306998 |
| Title : Immunochemical characterization of anti-acetylcholinesterase inhibitory monoclonal antibodies - Gentry_1993_Chem.Biol.Interact_87_227 |
| Author(s) : Gentry MK , Saxena A , Ashani Y , Doctor BP |
| Ref : Chemico-Biological Interactions , 87 :227 , 1993 |
| Abstract : Gentry_1993_Chem.Biol.Interact_87_227 |
| ESTHER : Gentry_1993_Chem.Biol.Interact_87_227 |
| PubMedSearch : Gentry_1993_Chem.Biol.Interact_87_227 |
| PubMedID: 7688272 |
| Title : Correlation of the anticholinesterase activity of a series of organophosphates with their ability to compete with agonist binding to muscarinic receptors - Ward_1993_Toxicol.Appl.Pharmacol_122_300 |
| Author(s) : Ward TR , Ferris DJ , Tilson HA , Mundy WR |
| Ref : Toxicology & Applied Pharmacology , 122 :300 , 1993 |
| Abstract : Ward_1993_Toxicol.Appl.Pharmacol_122_300 |
| ESTHER : Ward_1993_Toxicol.Appl.Pharmacol_122_300 |
| PubMedSearch : Ward_1993_Toxicol.Appl.Pharmacol_122_300 |
| PubMedID: 8212012 |
| Title : Therapeutic efficacy of bis-pyridinium oximes against diisopropyl fluorophosphate (DFP) and soman intoxication in rodents - |
| Author(s) : Dube SN , Kumar D , Sikder AK , Sikder N , Jaiswal DK , Das Gupta S |
| Ref : Pharmazie , 47 :68 , 1992 |
| PubMedID: 1608992 |
| Title : In vitro protection of acetylcholinesterase and butyrylcholinesterase by tetrahydroaminoacridine. Comparison with physostigmine - Galli_1992_Biochem.Pharmacol_43_2427 |
| Author(s) : Galli A , Mori F , Gori I , Lucherini M |
| Ref : Biochemical Pharmacology , 43 :2427 , 1992 |
| Abstract : Galli_1992_Biochem.Pharmacol_43_2427 |
| ESTHER : Galli_1992_Biochem.Pharmacol_43_2427 |
| PubMedSearch : Galli_1992_Biochem.Pharmacol_43_2427 |
| PubMedID: 1610407 |
| Title : Cholinesterases of heart muscle. Characterization of multiple enzymes using kinetics of irreversible organophosphorus inhibition - Chemnitius_1992_Biochem.Pharmacol_43_823 |
| Author(s) : Chemnitius JM , Chemnitius GC , Haselmeyer KH , Kreuzer H , Zech R |
| Ref : Biochemical Pharmacology , 43 :823 , 1992 |
| Abstract : Chemnitius_1992_Biochem.Pharmacol_43_823 |
| ESTHER : Chemnitius_1992_Biochem.Pharmacol_43_823 |
| PubMedSearch : Chemnitius_1992_Biochem.Pharmacol_43_823 |
| PubMedID: 1540236 |
| Title : Acylpeptide hydrolase: inhibitors and some active site residues of the human enzyme - Scaloni_1992_J.Biol.Chem_267_3811 |
| Author(s) : Scaloni A , Jones WM , Barra D , Pospischil M , Sassa S , Popowicz AM , Manning LR , Schneewind O , Manning JM |
| Ref : Journal of Biological Chemistry , 267 :3811 , 1992 |
| Abstract : Scaloni_1992_J.Biol.Chem_267_3811 |
| ESTHER : Scaloni_1992_J.Biol.Chem_267_3811 |
| PubMedSearch : Scaloni_1992_J.Biol.Chem_267_3811 |
| PubMedID: 1740429 |
| Title : Isolation of a tripeptide (Ala-Gly-Ser) exhibiting weak acetylthiocholine hydrolyzing activity from a high-salt soluble form of monkey diaphragm acetylcholinesterase - Jayanthi_1992_Neurochem.Res_17_351 |
| Author(s) : Jayanthi LD , Balasubramanian AS |
| Ref : Neurochemical Research , 17 :351 , 1992 |
| Abstract : Jayanthi_1992_Neurochem.Res_17_351 |
| ESTHER : Jayanthi_1992_Neurochem.Res_17_351 |
| PubMedSearch : Jayanthi_1992_Neurochem.Res_17_351 |
| PubMedID: 1513418 |
| Title : Hypothermia: limited tolerance to repeated soman administration and cross-tolerance to oxotremorine - Clement_1991_Pharmacol.Biochem.Behav_39_305 |
| Author(s) : Clement JG |
| Ref : Pharmacol Biochem Behav , 39 :305 , 1991 |
| Abstract : Clement_1991_Pharmacol.Biochem.Behav_39_305 |
| ESTHER : Clement_1991_Pharmacol.Biochem.Behav_39_305 |
| PubMedSearch : Clement_1991_Pharmacol.Biochem.Behav_39_305 |
| PubMedID: 1946573 |
| Title : A new H-oxime restores rat diaphragm contractility after esterase inhibition in vitro - Alberts_1990_Eur.J.Pharmacol_184_191 |
| Author(s) : Alberts P |
| Ref : European Journal of Pharmacology , 184 :191 , 1990 |
| Abstract : Alberts_1990_Eur.J.Pharmacol_184_191 |
| ESTHER : Alberts_1990_Eur.J.Pharmacol_184_191 |
| PubMedSearch : Alberts_1990_Eur.J.Pharmacol_184_191 |
| PubMedID: 2209712 |
| Title : Inhibition of cholinesterases by DFP and induction of organophosphate-detoxicating enzymes in rats - Kaliste-Korhonen_1990_Gen.Pharmacol_21_527 |
| Author(s) : Kaliste-Korhonen E , Torronen R , Ylitalo P , Hanninen O |
| Ref : General Pharmacology , 21 :527 , 1990 |
| Abstract : Kaliste-Korhonen_1990_Gen.Pharmacol_21_527 |
| ESTHER : Kaliste-Korhonen_1990_Gen.Pharmacol_21_527 |
| PubMedSearch : Kaliste-Korhonen_1990_Gen.Pharmacol_21_527 |
| PubMedID: 2165959 |
| Title : Repeated exposure to diisopropylfluorophosphate (DFP) produces increased sensitivity to cholinergic antagonists in discrimination retention and reversal - Raffaele_1990_Psychopharmacology_100_267 |
| Author(s) : Raffaele K , Olton D , Annau Z |
| Ref : Psychopharmacology , 100 :267 , 1990 |
| Abstract : Raffaele_1990_Psychopharmacology_100_267 |
| ESTHER : Raffaele_1990_Psychopharmacology_100_267 |
| PubMedSearch : Raffaele_1990_Psychopharmacology_100_267 |
| PubMedID: 2305015 |
| Title : Down-regulation of muscarinic receptors in the striatum of organophosphate-treated swine - Yang_1990_Toxicol.Appl.Pharmacol_104_375 |
| Author(s) : Yang CM , Dwyer TM , Mohan PM , Ho IK , Farley JM |
| Ref : Toxicol Appl Pharmacol , 104 :375 , 1990 |
| Abstract : Yang_1990_Toxicol.Appl.Pharmacol_104_375 |
| ESTHER : Yang_1990_Toxicol.Appl.Pharmacol_104_375 |
| PubMedSearch : Yang_1990_Toxicol.Appl.Pharmacol_104_375 |
| PubMedID: 2385834 |
| Title : [Catalytic properties of cholinesterases immobilized in a gelatin membrane] - Kuznetsova_1990_Ukr.Biokhim.Zh_62_42 |
| Author(s) : Kuznetsova LP , Kugusheva LI , Nikol'skaia EB |
| Ref : Ukr Biokhim Zh , 62 :42 , 1990 |
| Abstract : Kuznetsova_1990_Ukr.Biokhim.Zh_62_42 |
| ESTHER : Kuznetsova_1990_Ukr.Biokhim.Zh_62_42 |
| PubMedSearch : Kuznetsova_1990_Ukr.Biokhim.Zh_62_42 |
| PubMedID: 2087792 |
| Title : Cold exposure decreases the effectiveness of atropine-oxime treatment in organophosphate intoxication in rats and mice - Kaliste-Korhonen_1989_Gen.Pharmacol_20_805 |
| Author(s) : Kaliste-Korhonen E , Ryhanen R , Ylitalo P , Hanninen O |
| Ref : General Pharmacology , 20 :805 , 1989 |
| Abstract : Kaliste-Korhonen_1989_Gen.Pharmacol_20_805 |
| ESTHER : Kaliste-Korhonen_1989_Gen.Pharmacol_20_805 |
| PubMedSearch : Kaliste-Korhonen_1989_Gen.Pharmacol_20_805 |
| PubMedID: 2687080 |
| Title : Comparison of the effects of diisopropylfluorophosphate, sarin, soman, and tabun on toxicity and brain acetylcholinesterase activity in mice - Tripathi_1989_J.Toxicol.Environ.Health_26_437 |
| Author(s) : Tripathi HL , Dewey WL |
| Ref : J Toxicol Environ Health , 26 :437 , 1989 |
| Abstract : Tripathi_1989_J.Toxicol.Environ.Health_26_437 |
| ESTHER : Tripathi_1989_J.Toxicol.Environ.Health_26_437 |
| PubMedSearch : Tripathi_1989_J.Toxicol.Environ.Health_26_437 |
| PubMedID: 2709438 |
| Title : A microassay-based procedure for measuring low levels of toxic organophosphorus compounds through acetylcholinesterase inhibition - Hammond_1989_Anal.Biochem_180_380 |
| Author(s) : Hammond PS , Forster JS |
| Ref : Analytical Biochemistry , 180 :380 , 1989 |
| Abstract : Hammond_1989_Anal.Biochem_180_380 |
| ESTHER : Hammond_1989_Anal.Biochem_180_380 |
| PubMedSearch : Hammond_1989_Anal.Biochem_180_380 |
| PubMedID: 2817369 |
| Title : Toxicity of an acute dose of agent VX and other organophosphorus esters in the chicken - Wilson_1988_J.Toxicol.Environ.Health_23_103 |
| Author(s) : Wilson BW , Henderson JD , Chow E , Schreider J , Goldman M , Culbertson R , Dacre JC |
| Ref : J Toxicol Environ Health , 23 :103 , 1988 |
| Abstract : Wilson_1988_J.Toxicol.Environ.Health_23_103 |
| ESTHER : Wilson_1988_J.Toxicol.Environ.Health_23_103 |
| PubMedSearch : Wilson_1988_J.Toxicol.Environ.Health_23_103 |
| PubMedID: 3336055 |
| Title : Effect of the cold environment on organophosphate toxicity and inhibition of cholinesterase activity - Ryhanen_1988_Gen.Pharmacol_19_741 |
| Author(s) : Ryhanen R , Honkakoski P , Harri M , Ylitalo P , Hanninen O |
| Ref : General Pharmacology , 19 :741 , 1988 |
| Abstract : Ryhanen_1988_Gen.Pharmacol_19_741 |
| ESTHER : Ryhanen_1988_Gen.Pharmacol_19_741 |
| PubMedSearch : Ryhanen_1988_Gen.Pharmacol_19_741 |
| PubMedID: 3215484 |
| Title : Effects of the organophosphate cholinesterase inhibitor (DFP) on hypothalamic acetylcholinesterase and pituitary-adrenal activity - |
| Author(s) : Kokka N , Clemons GK , Lomax P |
| Ref : Proc West Pharmacol Soc , 30 :219 , 1987 |
| PubMedID: 3628281 |
| Title : A peptidase activity exhibited by human serum pseudocholinesterase - Boopathy_1987_Eur.J.Biochem_162_191 |
| Author(s) : Boopathy R , Balasubramanian AS |
| Ref : European Journal of Biochemistry , 162 :191 , 1987 |
| Abstract : Boopathy_1987_Eur.J.Biochem_162_191 |
| ESTHER : Boopathy_1987_Eur.J.Biochem_162_191 |
| PubMedSearch : Boopathy_1987_Eur.J.Biochem_162_191 |
| PubMedID: 3545820 |
| Title : Kinetic investigations into the interactions of aprophen with cholinesterases and a carboxylesterase - Rush_1986_Biochem.Pharmacol_35_4167 |
| Author(s) : Rush RS , Doctor BP , Wolfe AD |
| Ref : Biochemical Pharmacology , 35 :4167 , 1986 |
| Abstract : Rush_1986_Biochem.Pharmacol_35_4167 |
| ESTHER : Rush_1986_Biochem.Pharmacol_35_4167 |
| PubMedSearch : Rush_1986_Biochem.Pharmacol_35_4167 |
| PubMedID: 3098245 |
| Title : In-vitro and in-vivo protection of acetylcholinesterase by eseroline against inactivation by diisopropyl fluorophosphate and carbamates - Galli_1985_J.Pharm.Pharmacol_37_42 |
| Author(s) : Galli A , Malmberg-Aiello P , Renzi G , Bartolini A |
| Ref : J Pharm Pharmacol , 37 :42 , 1985 |
| Abstract : Galli_1985_J.Pharm.Pharmacol_37_42 |
| ESTHER : Galli_1985_J.Pharm.Pharmacol_37_42 |
| PubMedSearch : Galli_1985_J.Pharm.Pharmacol_37_42 |
| PubMedID: 2858526 |
| Title : Polymorphism of pseudocholinesterase in Torpedo marmorata tissues: comparative study of the catalytic and molecular properties of this enzyme with acetylcholinesterase - Toutant_1985_J.Neurochem_44_580 |
| Author(s) : Toutant JP , Massoulie J , Bon S |
| Ref : Journal of Neurochemistry , 44 :580 , 1985 |
| Abstract : Toutant_1985_J.Neurochem_44_580 |
| ESTHER : Toutant_1985_J.Neurochem_44_580 |
| PubMedSearch : Toutant_1985_J.Neurochem_44_580 |
| PubMedID: 2578181 |
| Title : Mechanisms involved in the development of tolerance to DFP toxicity - Gupta_1985_Fundam.Appl.Toxicol_5_S17 |
| Author(s) : Gupta RC , Patterson GT , Dettbarn WD |
| Ref : Fundamental & Applied Toxicology , 5 :S17 , 1985 |
| Abstract : Gupta_1985_Fundam.Appl.Toxicol_5_S17 |
| ESTHER : Gupta_1985_Fundam.Appl.Toxicol_5_S17 |
| PubMedSearch : Gupta_1985_Fundam.Appl.Toxicol_5_S17 |
| PubMedID: 4092885 |
| Title : Brain cholinesterases. Differentiation of target enzymes for toxic organophosphorus compounds - Chemnitius_1983_Biochem.Pharmacol_32_1693 |
| Author(s) : Chemnitius JM , Haselmeyer KH , Zech R |
| Ref : Biochemical Pharmacology , 32 :1693 , 1983 |
| Abstract : Chemnitius_1983_Biochem.Pharmacol_32_1693 |
| ESTHER : Chemnitius_1983_Biochem.Pharmacol_32_1693 |
| PubMedSearch : Chemnitius_1983_Biochem.Pharmacol_32_1693 |
| PubMedID: 6870909 |
| Title : Differential effects of di-isopropylfluorophosphate poisoning and its treatment on opioid antinociception in the mouse - Kitchen_1983_Life.Sci_1_669 |
| Author(s) : Kitchen I , Green PG |
| Ref : Life Sciences , 1 :669 , 1983 |
| Abstract : Kitchen_1983_Life.Sci_1_669 |
| ESTHER : Kitchen_1983_Life.Sci_1_669 |
| PubMedSearch : Kitchen_1983_Life.Sci_1_669 |
| PubMedID: 6664242 |
| Title : Evaluation of cytotoxic responses caused by selected organophosphorus esters in chick sympathetic ganglia cultures - Obersteiner_1978_Can.J.Comp.Med_42_80 |
| Author(s) : Obersteiner EJ , Sharma RP |
| Ref : Can J Comp Med , 42 :80 , 1978 |
| Abstract : Obersteiner_1978_Can.J.Comp.Med_42_80 |
| ESTHER : Obersteiner_1978_Can.J.Comp.Med_42_80 |
| PubMedSearch : Obersteiner_1978_Can.J.Comp.Med_42_80 |
| PubMedID: 565668 |
| Title : A radiometric study of cholinesterase and its inhibition - |
| Author(s) : Winteringham FP , Disney RW |
| Ref : Biochemical Journal , 91 :506 , 1964 |
| PubMedID: 5840710 |