DMBMP-thiocholine
can be synthetized as (Sp) or (P-) and (Rp) or (P+) stereoisomers
General
Type : Organophosphate,Trimethylammonium,Sulfur Compound
Chemical_Nomenclature : 2-(3,3-dimethylbutoxy-methyl-oxidophosphaniumyl)sulfanylethyl-trimethylazanium
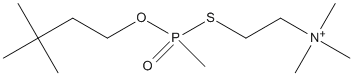
Canonical SMILES : CC(C)(C)CCO[P+](C)([O-])SCC[N+](C)(C)C
InChI : InChI=1S\/C12H29NO2PS\/c1-12(2,3)8-10-15-16(7,14)17-11-9-13(4,5)6\/h8-11H2,1-7H3\/q+1
InChIKey : BARGDJKEDLFIBS-UHFFFAOYSA-N
Other name(s) : 3,3-dimethylbutyl methylphosphonyl thiocholine
MW : 282.40
Formula : C12H29NO2PS+
CAS_number :
PubChem : 101255752
UniChem : BARGDJKEDLFIBS-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
References (3)
| Title : Mechanism of oxime reactivation of acetylcholinesterase analyzed by chirality and mutagenesis - Wong_2000_Biochemistry_39_5750 |
| Author(s) : Wong L , Radic Z , Bruggemann RJ , Hosea NA , Berman HA , Taylor P |
| Ref : Biochemistry , 39 :5750 , 2000 |
| Abstract : Wong_2000_Biochemistry_39_5750 |
| ESTHER : Wong_2000_Biochemistry_39_5750 |
| PubMedSearch : Wong_2000_Biochemistry_39_5750 |
| PubMedID: 10801325 |
| Title : Chiral reactions of acetylcholinesterase probed with enantiomeric methylphosphonothioates. Noncovalent determinants of enzyme chirality [published erratum appears in J Biol Chem 1989 Nov 25\;264(33):20154] - Berman_1989_J.Biol.Chem_264_3942 |
| Author(s) : Berman HA , Leonard K |
| Ref : Journal of Biological Chemistry , 264 :3942 , 1989 |
| Abstract : Berman_1989_J.Biol.Chem_264_3942 |
| ESTHER : Berman_1989_J.Biol.Chem_264_3942 |
| PubMedSearch : Berman_1989_J.Biol.Chem_264_3942 |
| PubMedID: 2917983 |
| Title : Chiral nature of covalent methylphosphonyl conjugates of acetylcholinesterase - Berman_1989_J.Biol.Chem_264_3951 |
| Author(s) : Berman HA , Decker MM |
| Ref : Journal of Biological Chemistry , 264 :3951 , 1989 |
| Abstract : Berman_1989_J.Biol.Chem_264_3951 |
| ESTHER : Berman_1989_J.Biol.Chem_264_3951 |
| PubMedSearch : Berman_1989_J.Biol.Chem_264_3951 |
| PubMedID: 2917984 |