Eseroline
An opioid agonist structurally related to physostigmine (eserine) and morphine. Metabolite of Physostigmine/Eserine. A portion of administered cymserine is metabolized in the body into eseroline, a potent mu opioid agonist and neurotoxin
General
Type : Derivative of physostigmine-eserine,Indole
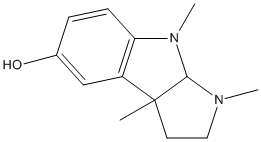
Chemical_Nomenclature : (3aS,8aR)-1,2,3,3a,8,8a-hexahydro-1,3a,8 trimethylpyrrolo[2,3-b]indo l-5-ol
Canonical SMILES : CC12CCN(C1N(C3=C2C=C(C=C3)O)C)C
InChI : InChI=1S\/C13H18N2O\/c1-13-6-7-14(2)12(13)15(3)11-5-4-9(16)8-10(11)13\/h4-5,8,12,16H,6-7H2,1-3H3 || InChI=1S\/C13H18N2O\/c1-13-6-7-14(2)12(13)15(3)11-5-4-9(16)8-10(11)13\/h4-5,8,12,16H,6-7H2,1-3H3\/t12-,13+\/m1\/s1
InChIKey : HKGWQUVGHPDEBZ-UHFFFAOYSA-N || HKGWQUVGHPDEBZ-OLZOCXBDSA-N
Other name(s) : CHEMBL310934,CHEBI:48845,ST057526,(3as-cis)-1,2,3,3a,8,8a-hexahydro-1,3a,8-trimethylpyrrolo[2,3-b]indol-5-ol,STK683285,AC1Q7A0V,SCHEMBL150459,AC1L1B22,CHEMBL2322559
MW : 218.295
Formula : C13H18N2O
CAS_number : 469-22-7
UniChem : HKGWQUVGHPDEBZ-UHFFFAOYSA-N || HKGWQUVGHPDEBZ-OLZOCXBDSA-N
IUPHAR :
Wikipedia : Eseroline
Target
References (9)
| Title : Reversible inhibition of cholinesterases by opioids: possible pharmacological consequences - Galli_1996_J.Pharm.Pharmacol_48_1164 |
| Author(s) : Galli A , Ranaudo E , Giannini L , Costagli C |
| Ref : J Pharm Pharmacol , 48 :1164 , 1996 |
| Abstract : Galli_1996_J.Pharm.Pharmacol_48_1164 |
| ESTHER : Galli_1996_J.Pharm.Pharmacol_48_1164 |
| PubMedSearch : Galli_1996_J.Pharm.Pharmacol_48_1164 |
| PubMedID: 8961166 |
| Title : Eseroline, a metabolite of physostigmine, induces neuronal cell death - Somani_1990_Toxicol.Appl.Pharmacol_106_28 |
| Author(s) : Somani SM , Kutty RK , Krishna G |
| Ref : Toxicol Appl Pharmacol , 106 :28 , 1990 |
| Abstract : Somani_1990_Toxicol.Appl.Pharmacol_106_28 |
| ESTHER : Somani_1990_Toxicol.Appl.Pharmacol_106_28 |
| PubMedSearch : Somani_1990_Toxicol.Appl.Pharmacol_106_28 |
| PubMedID: 2251681 |
| Title : Pharmacokinetics and pharmacodynamics of physostigmine in the rat after oral administration - Somani_1989_Biopharm.Drug.Dispos_10_187 |
| Author(s) : Somani SM |
| Ref : Biopharmaceutics & Drug Disposition , 10 :187 , 1989 |
| Abstract : Somani_1989_Biopharm.Drug.Dispos_10_187 |
| ESTHER : Somani_1989_Biopharm.Drug.Dispos_10_187 |
| PubMedSearch : Somani_1989_Biopharm.Drug.Dispos_10_187 |
| PubMedID: 2706318 |
| Title : Comparative inhibitory effects of various physostigmine analogs against acetyl- and butyrylcholinesterases - Atack_1989_J.Pharmacol.Exp.Ther_249_194 |
| Author(s) : Atack JR , Yu QS , Soncrant TT , Brossi A , Rapoport SI |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 249 :194 , 1989 |
| Abstract : Atack_1989_J.Pharmacol.Exp.Ther_249_194 |
| ESTHER : Atack_1989_J.Pharmacol.Exp.Ther_249_194 |
| PubMedSearch : Atack_1989_J.Pharmacol.Exp.Ther_249_194 |
| PubMedID: 2709330 |
| Title : Carbamate analogues of (-)-physostigmine: in vitro inhibition of acetyl- and butyrylcholinesterase - Yu_1988_FEBS.Lett_234_127 |
| Author(s) : Yu QS , Atack JR , Rapoport SI , Brossi A |
| Ref : FEBS Letters , 234 :127 , 1988 |
| Abstract : Yu_1988_FEBS.Lett_234_127 |
| ESTHER : Yu_1988_FEBS.Lett_234_127 |
| PubMedSearch : Yu_1988_FEBS.Lett_234_127 |
| PubMedID: 3391264 |
| Title : Pharmacokinetics and pharmacodynamics of physostigmine in the rat after intravenous administration - Somani_1987_Drug.Metab.Dispos_15_627 |
| Author(s) : Somani SM , Khalique A |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 15 :627 , 1987 |
| Abstract : Somani_1987_Drug.Metab.Dispos_15_627 |
| ESTHER : Somani_1987_Drug.Metab.Dispos_15_627 |
| PubMedSearch : Somani_1987_Drug.Metab.Dispos_15_627 |
| PubMedID: 2891478 |
| Title : Distribution and pharmacokinetics of physostigmine in rat after intramuscular administration - Somani_1986_Fundam.Appl.Toxicol_6_327 |
| Author(s) : Somani SM , Khalique A |
| Ref : Fundamental & Applied Toxicology , 6 :327 , 1986 |
| Abstract : Somani_1986_Fundam.Appl.Toxicol_6_327 |
| ESTHER : Somani_1986_Fundam.Appl.Toxicol_6_327 |
| PubMedSearch : Somani_1986_Fundam.Appl.Toxicol_6_327 |
| PubMedID: 3699321 |
| Title : In-vitro and in-vivo protection of acetylcholinesterase by eseroline against inactivation by diisopropyl fluorophosphate and carbamates - Galli_1985_J.Pharm.Pharmacol_37_42 |
| Author(s) : Galli A , Malmberg-Aiello P , Renzi G , Bartolini A |
| Ref : J Pharm Pharmacol , 37 :42 , 1985 |
| Abstract : Galli_1985_J.Pharm.Pharmacol_37_42 |
| ESTHER : Galli_1985_J.Pharm.Pharmacol_37_42 |
| PubMedSearch : Galli_1985_J.Pharm.Pharmacol_37_42 |
| PubMedID: 2858526 |
| Title : Reversible inhibition of acetylcholinesterase by eseroline, an opioid agonist structurally related to physostigmine (eserine) and morphine - Galli_1982_Biochem.Pharmacol_31_1233 |
| Author(s) : Galli A , Renzi G , Grazzini E , Bartolini R , Malmberg-Aiello P , Bartolini A |
| Ref : Biochemical Pharmacology , 31 :1233 , 1982 |
| Abstract : Galli_1982_Biochem.Pharmacol_31_1233 |
| ESTHER : Galli_1982_Biochem.Pharmacol_31_1233 |
| PubMedSearch : Galli_1982_Biochem.Pharmacol_31_1233 |
| PubMedID: 7092918 |