GSK2256294A
inhibitor of Epoxide hydrolase developed by GlaxoSmithKline as investigational new drug (IND) candidate that has reached Phase I clinical trials for COPD . has not progressed to further stages of clinical trials
General
Type : Urea derivative,Cyanide,Carboxamide,Triazine
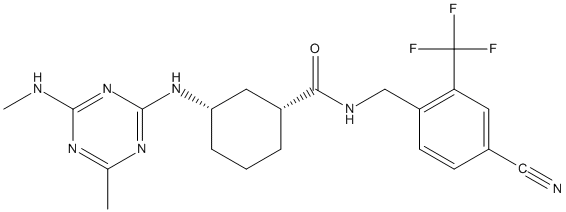
Chemical_Nomenclature : (1R,3S)-N-[[4-cyano-2-(trifluoromethyl)phenyl]methyl]-3-[[4-methyl-6-(methylamino)-1,3,5-triazin-2-yl]amino]cyclohexane-1-carboxamide
Canonical SMILES : CC1=NC(=NC(=N1)NC)NC2CCCC(C2)C(=O)NCC3=C(C=C(C=C3)C#N)C(F)(F)F
InChI : InChI=1S\/C21H24F3N7O\/c1-12-28-19(26-2)31-20(29-12)30-16-5-3-4-14(9-16)18(32)27-11-15-7-6-13(10-25)8-17(15)21(22,23)24\/h6-8,14,16H,3-5,9,11H2,1-2H3,(H,27,32)(H2,26,28,29,30,31)\/t14-,16+\/m1\/s1
InChIKey : LQHDJQIMETZMPH-ZBFHGGJFSA-N
Other name(s) : UNII-L33EX3XR0T,GSK-2256294,GSK 2256294A,L33EX3XR0T,MolPort-042-665-838,CHEMBL3818875
MW : 447.46
Formula : C21H24F3N7O
CAS_number : 1142090-23-0
PubChem : 59448236
UniChem : LQHDJQIMETZMPH-ZBFHGGJFSA-N
IUPHAR :
Wikipedia :
Target
Families : GSK2256294A ligand of proteins in family: Epoxide_hydrolase
Stucture :
Protein : human-EPHX2
References (8)
| Title : Discovery of soluble epoxide hydrolase inhibitors through DNA-encoded library technology (ELT) - Ding_2021_Bioorg.Med.Chem_41_116216 |
| Author(s) : Ding Y , Belyanskaya S , DeLorey JL , Messer JA , Joseph Franklin G , Centrella PA , Morgan BA , Clark MA , Skinner SR , Dodson JW , Li P , Marino JP, Jr. , Israel DI |
| Ref : Bioorganic & Medicinal Chemistry , 41 :116216 , 2021 |
| Abstract : Ding_2021_Bioorg.Med.Chem_41_116216 |
| ESTHER : Ding_2021_Bioorg.Med.Chem_41_116216 |
| PubMedSearch : Ding_2021_Bioorg.Med.Chem_41_116216 |
| PubMedID: 34023664 |
| Title : The soluble epoxide hydrolase inhibitor GSK2256294 decreases the proportion of adipose pro-inflammatory T cells - Mashayekhi_2021_Prostaglandins.Other.Lipid.Mediat_158_106604 |
| Author(s) : Mashayekhi M , Wanjalla CN , Warren CM , Simmons JD , Ghoshal K , Pilkinton M , Bailin SS , Gabriel CL , Pozzi A , Koethe JR , Brown NJ , Kalams SA , Luther JM |
| Ref : Prostaglandins Other Lipid Mediat , 158 :106604 , 2021 |
| Abstract : Mashayekhi_2021_Prostaglandins.Other.Lipid.Mediat_158_106604 |
| ESTHER : Mashayekhi_2021_Prostaglandins.Other.Lipid.Mediat_158_106604 |
| PubMedSearch : Mashayekhi_2021_Prostaglandins.Other.Lipid.Mediat_158_106604 |
| PubMedID: 34922004 |
| Title : GSK2256294 Decreases sEH (Soluble Epoxide Hydrolase) Activity in Plasma, Muscle, and Adipose and Reduces F2-Isoprostanes but Does Not Alter Insulin Sensitivity in Humans - |
| Author(s) : Luther JM , Ray J , Wei D , Koethe JR , Hannah L , DeMatteo A , Manning R , Terker AS , Peng D , Nian H , Yu C , Mashayekhi M , Gamboa J , Brown NJ |
| Ref : Hypertension , 78 :1092 , 2021 |
| PubMedID: 34455816 |
| Title : Identification and optimization of soluble epoxide hydrolase inhibitors with dual potency towards fatty acid amide hydrolase - Kodani_2018_Bioorg.Med.Chem.Lett_28_762 |
| Author(s) : Kodani SD , Bhakta S , Hwang SH , Pakhomova S , Newcomer ME , Morisseau C , Hammock BD |
| Ref : Bioorganic & Medicinal Chemistry Lett , 28 :762 , 2018 |
| Abstract : Kodani_2018_Bioorg.Med.Chem.Lett_28_762 |
| ESTHER : Kodani_2018_Bioorg.Med.Chem.Lett_28_762 |
| PubMedSearch : Kodani_2018_Bioorg.Med.Chem.Lett_28_762 |
| PubMedID: 29366648 |
| Gene_locus related to this paper: human-EPHX2 |
| Title : Mechanisms of Vascular Dysfunction in COPD and Effects of a Novel Soluble Epoxide Hydrolase Inhibitor in Smokers - Yang_2017_Chest_151_555 |
| Author(s) : Yang L , Cheriyan J , Gutterman DD , Mayer RJ , Ament Z , Griffin JL , Lazaar AL , Newby DE , Tal-Singer R , Wilkinson IB |
| Ref : Chest , 151 :555 , 2017 |
| Abstract : Yang_2017_Chest_151_555 |
| ESTHER : Yang_2017_Chest_151_555 |
| PubMedSearch : Yang_2017_Chest_151_555 |
| PubMedID: 27884766 |
| Title : Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor - Lazaar_2016_Br.J.Clin.Pharmacol_81_971 |
| Author(s) : Lazaar AL , Yang L , Boardley RL , Goyal NS , Robertson J , Baldwin SJ , Newby DE , Wilkinson IB , Tal-Singer R , Mayer RJ , Cheriyan J |
| Ref : British Journal of Clinical Pharmacology , 81 :971 , 2016 |
| Abstract : Lazaar_2016_Br.J.Clin.Pharmacol_81_971 |
| ESTHER : Lazaar_2016_Br.J.Clin.Pharmacol_81_971 |
| PubMedSearch : Lazaar_2016_Br.J.Clin.Pharmacol_81_971 |
| PubMedID: 26620151 |
| Title : In vitro and in vivo characterization of a novel soluble epoxide hydrolase inhibitor - Podolin_2013_Prostaglandins.Other.Lipid.Mediat_104-105_25 |
| Author(s) : Podolin PL , Bolognese BJ , Foley JF , Long E, 3rd , Peck B , Umbrecht S , Zhang X , Zhu P , Schwartz B , Xie W , Quinn C , Qi H , Sweitzer S , Chen S , Galop M , Ding Y , Belyanskaya SL , Israel DI , Morgan BA , Behm DJ , Marino JP, Jr. , Kurali E , Barnette MS , Mayer RJ , Booth-Genthe CL , Callahan JF |
| Ref : Prostaglandins Other Lipid Mediat , 104-105 :25 , 2013 |
| Abstract : Podolin_2013_Prostaglandins.Other.Lipid.Mediat_104-105_25 |
| ESTHER : Podolin_2013_Prostaglandins.Other.Lipid.Mediat_104-105_25 |
| PubMedSearch : Podolin_2013_Prostaglandins.Other.Lipid.Mediat_104-105_25 |
| PubMedID: 23434473 |
| Title : Soluble epoxide hydrolase inhibition does not prevent cardiac remodeling and dysfunction after aortic constriction in rats and mice - Morgan_2013_J.Cardiovasc.Pharmacol_61_291 |
| Author(s) : Morgan LA , Olzinski AR , Upson JJ , Zhao S , Wang T , Eisennagel SH , Hoang B , Tunstead JR , Marino JP, Jr. , Willette RN , Jucker BM , Behm DJ |
| Ref : J Cardiovasc Pharmacol , 61 :291 , 2013 |
| Abstract : Morgan_2013_J.Cardiovasc.Pharmacol_61_291 |
| ESTHER : Morgan_2013_J.Cardiovasc.Pharmacol_61_291 |
| PubMedSearch : Morgan_2013_J.Cardiovasc.Pharmacol_61_291 |
| PubMedID: 23232840 |