Heptylphysostigmine
Treatment of Alzheimer Disease
General
Type : Derivative of physostigmine-eserine,Carbamate,Indole
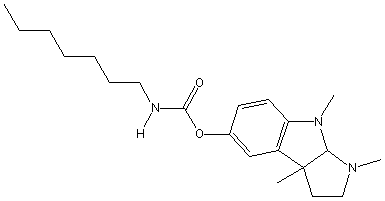
Chemical_Nomenclature : (3,4,8b-trimethyl-2,3a-dihydro-1H-pyrrolo[2,3-b]indol-7-yl) N-heptylcarbamate
Canonical SMILES : CCCCCCCNC(=O)OC1=CC2=C(C=C1)N(C3C2(CCN3C)C)C
InChI : InChI=1S\/C21H33N3O2\/c1-5-6-7-8-9-13-22-20(25)26-16-10-11-18-17(15-16)21(2)12-14-23(3)19(21)24(18)4\/h10-11,15,19H,5-9,12-14H2,1-4H3,(H,22,25)
InChIKey : RRGMXBQMCUKRLH-UHFFFAOYSA-N
Other name(s) : pyrrolo 2,3b indol-5-ol 3,3a,8,8a-hexahydro-1,3a,8-trimethylheptylcarbamate ester 3aS-cis,Heptylstigmine,Eptastigmine,Eseroline, heptylcarbamate,N-Demethyl-N-heptylphysostigmine,Eseroline, heptylcarbamate(ester),MF-201,MF201,heptastigmine,Eptastigmine, (Mediolanum)
MW : 359.506
Formula : C21H33N3O2
CAS_number : 101246-68-8
PubChem : 612747
UniChem : RRGMXBQMCUKRLH-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
References (29)
| Title : Invited review: Cholinesterase inhibitors for Alzheimer's disease therapy: from tacrine to future applications - Giacobini_1998_Neurochem.Int_32_413 |
| Author(s) : Giacobini E |
| Ref : Neurochem Int , 32 :413 , 1998 |
| Abstract : Giacobini_1998_Neurochem.Int_32_413 |
| ESTHER : Giacobini_1998_Neurochem.Int_32_413 |
| PubMedSearch : Giacobini_1998_Neurochem.Int_32_413 |
| PubMedID: 9676739 |
| Title : Inhibition of cholinesterase-associated aryl acylamidase activity by anticholinesterase agents: focus on drugs potentially effective in Alzheimer's disease - Costagli_1998_Biochem.Pharmacol_55_1733 |
| Author(s) : Costagli C , Galli A |
| Ref : Biochemical Pharmacology , 55 :1733 , 1998 |
| Abstract : Costagli_1998_Biochem.Pharmacol_55_1733 |
| ESTHER : Costagli_1998_Biochem.Pharmacol_55_1733 |
| PubMedSearch : Costagli_1998_Biochem.Pharmacol_55_1733 |
| PubMedID: 9634011 |
| Title : Effect of the subchronic treatment with the acetylcholinesterase inhibitor heptastigmine on central cholinergic transmission and memory impairment in aged rats - Garrone_1998_Neurosci.Lett_245_53 |
| Author(s) : Garrone B , Luparini MR , Tolu L , Magnani M , Landolfi C , Milanese C |
| Ref : Neuroscience Letters , 245 :53 , 1998 |
| Abstract : Garrone_1998_Neurosci.Lett_245_53 |
| ESTHER : Garrone_1998_Neurosci.Lett_245_53 |
| PubMedSearch : Garrone_1998_Neurosci.Lett_245_53 |
| PubMedID: 9596354 |
| Title : Effect of food on the absorption of eptastigmine - Bjornsson_1998_Eur.J.Clin.Pharmacol_54_243 |
| Author(s) : Bjornsson TD , Troetel WM , Imbimbo BP |
| Ref : European Journal of Clinical Pharmacology , 54 :243 , 1998 |
| Abstract : Bjornsson_1998_Eur.J.Clin.Pharmacol_54_243 |
| ESTHER : Bjornsson_1998_Eur.J.Clin.Pharmacol_54_243 |
| PubMedSearch : Bjornsson_1998_Eur.J.Clin.Pharmacol_54_243 |
| PubMedID: 9681667 |
| Title : Maximum tolerated dose and pharmacodynamics of eptastigmine in elderly healthy volunteers - Mant_1998_J.Clin.Pharmacol_38_610 |
| Author(s) : Mant T , Troetel WM , Imbimbo BP |
| Ref : Journal of Clinical Pharmacology , 38 :610 , 1998 |
| Abstract : Mant_1998_J.Clin.Pharmacol_38_610 |
| ESTHER : Mant_1998_J.Clin.Pharmacol_38_610 |
| PubMedSearch : Mant_1998_J.Clin.Pharmacol_38_610 |
| PubMedID: 9702845 |
| Title : Cholinesterase inhibition improves blood flow in the ischemic cerebral cortex - Scremin_1997_Brain.Res.Bull_42_59 |
| Author(s) : Scremin OU , Li MG , Scremin AM , Jenden DJ |
| Ref : Brain Research Bulletin , 42 :59 , 1997 |
| Abstract : Scremin_1997_Brain.Res.Bull_42_59 |
| ESTHER : Scremin_1997_Brain.Res.Bull_42_59 |
| PubMedSearch : Scremin_1997_Brain.Res.Bull_42_59 |
| PubMedID: 8978935 |
| Title : Cholinergic control of cerebral blood flow in stroke, trauma and aging - Scremin_1996_Life.Sci_58(22)_2011 |
| Author(s) : Scremin OU , Jenden DJ |
| Ref : Life Sciences , 58 :2011 , 1996 |
| Abstract : Scremin_1996_Life.Sci_58(22)_2011 |
| ESTHER : Scremin_1996_Life.Sci_58(22)_2011 |
| PubMedSearch : Scremin_1996_Life.Sci_58(22)_2011 |
| PubMedID: 8637431 |
| Title : Eptastigmine-phosphotriesterase combination in DFP intoxication - Tuovinen_1996_Toxicol.Appl.Pharmacol_140_364 |
| Author(s) : Tuovinen K , Kaliste-Korhonen E , Raushel FM , Hanninen O |
| Ref : Toxicology & Applied Pharmacology , 140 :364 , 1996 |
| Abstract : Tuovinen_1996_Toxicol.Appl.Pharmacol_140_364 |
| ESTHER : Tuovinen_1996_Toxicol.Appl.Pharmacol_140_364 |
| PubMedSearch : Tuovinen_1996_Toxicol.Appl.Pharmacol_140_364 |
| PubMedID: 8887453 |
| Title : Phosphotriesterase, pralidoxime-2-chloride (2-PAM) and eptastigmine treatments and their combinations in DFP intoxication - Tuovinen_1996_Toxicol.Appl.Pharmacol_141_555 |
| Author(s) : Tuovinen K , Kaliste-Korhonen E , Raushel FM , Hanninen O |
| Ref : Toxicology & Applied Pharmacology , 141 :555 , 1996 |
| Abstract : Tuovinen_1996_Toxicol.Appl.Pharmacol_141_555 |
| ESTHER : Tuovinen_1996_Toxicol.Appl.Pharmacol_141_555 |
| PubMedSearch : Tuovinen_1996_Toxicol.Appl.Pharmacol_141_555 |
| PubMedID: 8975781 |
| Title : An inverted U-shaped curve for heptylphysostigmine on radial maze performance in rats: comparison with other cholinesterase inhibitors - Braida_1996_Eur.J.Pharmacol_302_13 |
| Author(s) : Braida D , Paladini E , Griffini P , Lamperti M , Maggi A , Sala M |
| Ref : European Journal of Pharmacology , 302 :13 , 1996 |
| Abstract : Braida_1996_Eur.J.Pharmacol_302_13 |
| ESTHER : Braida_1996_Eur.J.Pharmacol_302_13 |
| PubMedSearch : Braida_1996_Eur.J.Pharmacol_302_13 |
| PubMedID: 8790986 |
| Title : A multiple-dose safety trial of eptastigmine in Alzheimer's disease, with pharmacodynamic observations of red blood cell cholinesterase - Sramek_1995_Life.Sci_56_319 |
| Author(s) : Sramek JJ , Block GA , Reines SA , Sawin SF , Barchowsky A , Cutler NR |
| Ref : Life Sciences , 56 :319 , 1995 |
| Abstract : Sramek_1995_Life.Sci_56_319 |
| ESTHER : Sramek_1995_Life.Sci_56_319 |
| PubMedSearch : Sramek_1995_Life.Sci_56_319 |
| PubMedID: 7837931 |
| Title : The behavioral effects of heptylphysostigmine on rats lesioned in the nucleus basalis - Waite_1995_Neurosci.Research_21_251 |
| Author(s) : Waite JJ , Thal LJ |
| Ref : Neuroscience Research , 21 :251 , 1995 |
| Abstract : Waite_1995_Neurosci.Research_21_251 |
| ESTHER : Waite_1995_Neurosci.Research_21_251 |
| PubMedSearch : Waite_1995_Neurosci.Research_21_251 |
| PubMedID: 7753506 |
| Title : Relationship between pharmacokinetics and pharmacodynamics of eptastigmine in young healthy volunteers - Imbimbo_1995_J.Clin.Pharmacol_35_285 |
| Author(s) : Imbimbo BP , Licini M , Schettino M , Mosca A , Onelli E , Zecca L , Giustina A |
| Ref : Journal of Clinical Pharmacology , 35 :285 , 1995 |
| Abstract : Imbimbo_1995_J.Clin.Pharmacol_35_285 |
| ESTHER : Imbimbo_1995_J.Clin.Pharmacol_35_285 |
| PubMedSearch : Imbimbo_1995_J.Clin.Pharmacol_35_285 |
| PubMedID: 7608318 |
| Title : A patient-side technique for real-time measurement of acetylcholinesterase activity during monitoring of eptastigmine treatment - Mosca_1995_Therapeut.Drug.Monit_17_230 |
| Author(s) : Mosca A , Onelli E , Rosti E , Paleari R , Luzzana M , Imbimbo BP |
| Ref : Therapeutic Drug Monitoring , 17 :230 , 1995 |
| Abstract : Mosca_1995_Therapeut.Drug.Monit_17_230 |
| ESTHER : Mosca_1995_Therapeut.Drug.Monit_17_230 |
| PubMedSearch : Mosca_1995_Therapeut.Drug.Monit_17_230 |
| PubMedID: 7624918 |
| Title : Clinical pharmacokinetics of drugs for Alzheimer's disease - Parnetti_1995_Clin.Pharmacokinet_29_110 |
| Author(s) : Parnetti L |
| Ref : Clinical Pharmacokinetics , 29 :110 , 1995 |
| Abstract : Parnetti_1995_Clin.Pharmacokinet_29_110 |
| ESTHER : Parnetti_1995_Clin.Pharmacokinet_29_110 |
| PubMedSearch : Parnetti_1995_Clin.Pharmacokinet_29_110 |
| PubMedID: 7586900 |
| Title : Cholinesterase inhibitor effects on neurotransmitters in rat cortex in vivo - Cuadra_1994_J.Pharmacol.Exp.Ther_270_277 |
| Author(s) : Cuadra G , Summers K , Giacobini E |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 270 :277 , 1994 |
| Abstract : Cuadra_1994_J.Pharmacol.Exp.Ther_270_277 |
| ESTHER : Cuadra_1994_J.Pharmacol.Exp.Ther_270_277 |
| PubMedSearch : Cuadra_1994_J.Pharmacol.Exp.Ther_270_277 |
| PubMedID: 7913496 |
| Title : Synthesis of 2'-heptylcarbamoyloxy-2-methyl-6,7-benzomorphan: a new analogue of heptylphysostigmine (MF 201) - Brufani_1994_Farmaco_40_743 |
| Author(s) : Brufani M , Filocamo L , Imbriani E , Lappa S , Mannina L |
| Ref : Farmaco , 40 :743 , 1994 |
| Abstract : Brufani_1994_Farmaco_40_743 |
| ESTHER : Brufani_1994_Farmaco_40_743 |
| PubMedSearch : Brufani_1994_Farmaco_40_743 |
| PubMedID: 7832976 |
| Title : The effects of novel cholinesterase inhibitors and selective muscarinic receptor agonists in tests of reference and working memory - Dawson_1993_Behav.Brain.Res_57_143 |
| Author(s) : Dawson GR , Iversen SD |
| Ref : Behavioural Brain Research , 57 :143 , 1993 |
| Abstract : Dawson_1993_Behav.Brain.Res_57_143 |
| ESTHER : Dawson_1993_Behav.Brain.Res_57_143 |
| PubMedSearch : Dawson_1993_Behav.Brain.Res_57_143 |
| PubMedID: 8117420 |
| Title : Prolonged effects of cholinesterase inhibition with eptastigmine on the cerebral blood flow-metabolism ratio of normal rats - Scremin_1993_J.Cereb.Blood.Flow.Metab_13_702 |
| Author(s) : Scremin OU , Scremin AM , Heuser D , Hudgell R , Romero E , Imbimbo BP |
| Ref : Journal of Cerebral Blood Flow & Metabolism , 13 :702 , 1993 |
| Abstract : Scremin_1993_J.Cereb.Blood.Flow.Metab_13_702 |
| ESTHER : Scremin_1993_J.Cereb.Blood.Flow.Metab_13_702 |
| PubMedSearch : Scremin_1993_J.Cereb.Blood.Flow.Metab_13_702 |
| PubMedID: 8314923 |
| Title : Cholinesterase inhibitor effects on extracellular acetylcholine in rat cortex - Messamore_1993_Neuropharmacol_32_745 |
| Author(s) : Messamore E , Warpman U , Ogane N , Giacobini E |
| Ref : Neuropharmacology , 32 :745 , 1993 |
| Abstract : Messamore_1993_Neuropharmacol_32_745 |
| ESTHER : Messamore_1993_Neuropharmacol_32_745 |
| PubMedSearch : Messamore_1993_Neuropharmacol_32_745 |
| PubMedID: 8413838 |
| Title : Transdermal patch delivery of acetylcholinesterase inhibitors - Moriearty_1993_Meth.Find.Exp.Clin.Pharmacol_15_407 |
| Author(s) : Moriearty PL , Thornton SL , Becker RE |
| Ref : Methods Find Exp Clin Pharmacol , 15 :407 , 1993 |
| Abstract : Moriearty_1993_Meth.Find.Exp.Clin.Pharmacol_15_407 |
| ESTHER : Moriearty_1993_Meth.Find.Exp.Clin.Pharmacol_15_407 |
| PubMedSearch : Moriearty_1993_Meth.Find.Exp.Clin.Pharmacol_15_407 |
| PubMedID: 8231460 |
| Title : Reversal of cognitive impairment by heptyl physostigmine, a long-lasting cholinesterase inhibitor, in primates - Rupniak_1992_J.Neurol.Sci_107_246 |
| Author(s) : Rupniak NM , Tye SJ , Brazell C , Heald A , Iversen SD , Pagella PG |
| Ref : Journal of Neurology Sci , 107 :246 , 1992 |
| Abstract : Rupniak_1992_J.Neurol.Sci_107_246 |
| ESTHER : Rupniak_1992_J.Neurol.Sci_107_246 |
| PubMedSearch : Rupniak_1992_J.Neurol.Sci_107_246 |
| PubMedID: 1564524 |
| Title : Pharmacokinetics of heptastigmine in rats - Segre_1992_Pharmacol.Res_25_139 |
| Author(s) : Segre G , Cerretani D , Baldi A , Urso R |
| Ref : Pharmacol Res , 25 :139 , 1992 |
| Abstract : Segre_1992_Pharmacol.Res_25_139 |
| ESTHER : Segre_1992_Pharmacol.Res_25_139 |
| PubMedSearch : Segre_1992_Pharmacol.Res_25_139 |
| PubMedID: 1635892 |
| Title : Heptyl-physostigmine enhances basal forebrain control of cortical cerebral blood flow - Linville_1992_J.Neurosci.Res_31_573 |
| Author(s) : Linville DG , Giacobini E , Arneric SP |
| Ref : Journal of Neuroscience Research , 31 :573 , 1992 |
| Abstract : Linville_1992_J.Neurosci.Res_31_573 |
| ESTHER : Linville_1992_J.Neurosci.Res_31_573 |
| PubMedSearch : Linville_1992_J.Neurosci.Res_31_573 |
| PubMedID: 1640506 |
| Title : Determination of picogram levels of heptylphysostigmine in human plasma using high-performance liquid chromatography with fluorescence detection - Herold_1992_J.Chromatogr_581_227 |
| Author(s) : Herold ML , Constanzer ML , Matuszewski BK |
| Ref : Journal of Chromatography , 581 :227 , 1992 |
| Abstract : Herold_1992_J.Chromatogr_581_227 |
| ESTHER : Herold_1992_J.Chromatogr_581_227 |
| PubMedSearch : Herold_1992_J.Chromatogr_581_227 |
| PubMedID: 1452613 |
| Title : Inhibition of human brain and RBC acetylcholinesterase (AChE) by heptylphysostigmine (HPTL) - Moriearty_1992_Meth.Find.Exp.Clin.Pharmacol_14_615 |
| Author(s) : Moriearty PL , Becker RE |
| Ref : Methods Find Exp Clin Pharmacol , 14 :615 , 1992 |
| Abstract : Moriearty_1992_Meth.Find.Exp.Clin.Pharmacol_14_615 |
| ESTHER : Moriearty_1992_Meth.Find.Exp.Clin.Pharmacol_14_615 |
| PubMedSearch : Moriearty_1992_Meth.Find.Exp.Clin.Pharmacol_14_615 |
| PubMedID: 1494302 |
| Title : Preferential inhibition of acetylcholinesterase molecular forms in rat brain - Ogane_1992_Neurochem.Res_17_489 |
| Author(s) : Ogane N , Giacobini E , Messamore E |
| Ref : Neurochemical Research , 17 :489 , 1992 |
| Abstract : Ogane_1992_Neurochem.Res_17_489 |
| ESTHER : Ogane_1992_Neurochem.Res_17_489 |
| PubMedSearch : Ogane_1992_Neurochem.Res_17_489 |
| PubMedID: 1528356 |
| Title : Kinetics of cholinesterase inhibition by eptastigmine in man [letter] - |
| Author(s) : Unni LK , Hutt V , Imbimbo BP , Becker RE |
| Ref : European Journal of Clinical Pharmacology , 41 :83 , 1991 |
| PubMedID: 1782985 |