HuperzineA
Agent in Chinese herbal medicine; used in treatment of dementia; potential for use in Alzheimer's disease. purified from Huperzia serrata (Thunb.) Trev [syn. Lycopodium serratum Thunb.(Qian ceng ta). The natural (-)-huperzine A is 38 fold more potent than its enantiomer (+)-huperzine A. Also low binding to the N-methyl-D-aspartate (NMDA) receptors
General
Type : Derivative of Huperzine,Natural,Alkaloid
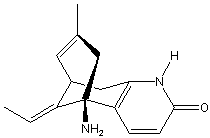
Chemical_Nomenclature : (5R,9R,11E)-5-amino-11-ethylidene-5,6.9,10-tetrahydro-7-methyl-5,9-methanocycloocteno[b]pyridin-2(1H)-one
Canonical SMILES : CC=C1C2CC3=C(C1(CC(=C2)C)N)C=CC(=O)N3
InChI : InChI=1S\/C15H18N2O\/c1-3-11-10-6-9(2)8-15(11,16)12-4-5-14(18)17-13(12)7-10\/h3-6,10H,7-8,16H2,1-2H3,(H,17,18)\/b11-3+\/t10-,15+\/m0\/s1
InChIKey : ZRJBHWIHUMBLCN-YQEJDHNASA-N
Other name(s) : Fordine,Huperzine,Huperzine A,(-)-huperzine A,CHEMBANK164,C-117,103735-86-0,HUP,Isomer of Selagine
MW : 242.3
Formula : C15H18N2O
CAS_number : 102518-79-6
PubChem : 854026
UniChem : ZRJBHWIHUMBLCN-YQEJDHNASA-N
IUPHAR :
Wikipedia : Huperzine
Target
Families : HuperzineA ligand of proteins in family: ACHE
Stucture :
Protein : torca-ACHE || human-ACHE
References (22)
| Title : Huperzine alkaloids: forty years of total syntheses - Cheng_2023_Nat.Prod.Rep__ |
| Author(s) : Cheng B , Song L , Chen F |
| Ref : Nat Prod Rep , : , 2023 |
| Abstract : Cheng_2023_Nat.Prod.Rep__ |
| ESTHER : Cheng_2023_Nat.Prod.Rep__ |
| PubMedSearch : Cheng_2023_Nat.Prod.Rep__ |
| PubMedID: 37818549 |
| Title : Structures of human acetylcholinesterase in complex with pharmacologically important ligands - Cheung_2012_J.Med.Chem_55_10282 |
| Author(s) : Cheung J , Rudolph MJ , Burshteyn F , Cassidy MS , Gary EN , Love J , Franklin MC , Height JJ |
| Ref : Journal of Medicinal Chemistry , 55 :10282 , 2012 |
| Abstract : Cheung_2012_J.Med.Chem_55_10282 |
| ESTHER : Cheung_2012_J.Med.Chem_55_10282 |
| PubMedSearch : Cheung_2012_J.Med.Chem_55_10282 |
| PubMedID: 23035744 |
| Gene_locus related to this paper: human-ACHE |
| Title : Improving effects of huperzine A on spatial working memory in aged monkeys and young adult monkeys with experimental cognitive impairment - Ye_1999_J.Pharmacol.Exp.Ther_288_814 |
| Author(s) : Ye JW , Cai JX , Wang LM , Tang XC |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 288 :814 , 1999 |
| Abstract : Ye_1999_J.Pharmacol.Exp.Ther_288_814 |
| ESTHER : Ye_1999_J.Pharmacol.Exp.Ther_288_814 |
| PubMedSearch : Ye_1999_J.Pharmacol.Exp.Ther_288_814 |
| PubMedID: 9918593 |
| Title : Synthesis and acetylcholinesterase inhibitory activity of (+\/-)-14- fluorohuperzine A - Zeng_1998_Bioorg.Med.Chem.Lett_8_1661 |
| Author(s) : Zeng F , Jiang H , Tang X , Chen K , Ji R |
| Ref : Bioorganic & Medicinal Chemistry Lett , 8 :1661 , 1998 |
| Abstract : Zeng_1998_Bioorg.Med.Chem.Lett_8_1661 |
| ESTHER : Zeng_1998_Bioorg.Med.Chem.Lett_8_1661 |
| PubMedSearch : Zeng_1998_Bioorg.Med.Chem.Lett_8_1661 |
| PubMedID: 9873409 |
| Title : Huperzine A--an interesting anticholinesterase compound from the Chinese herbal medicine - Patocka_1998_Acta.Medica_41_155 |
| Author(s) : Patocka J |
| Ref : Acta Medica , 41 :155 , 1998 |
| Abstract : Patocka_1998_Acta.Medica_41_155 |
| ESTHER : Patocka_1998_Acta.Medica_41_155 |
| PubMedSearch : Patocka_1998_Acta.Medica_41_155 |
| PubMedID: 9951045 |
| Title : Synthesis and evaluation of tacrine-huperzine A hybrids as acetylcholinesterase inhibitors of potential interest for the treatment of Alzheimer's disease - Badia_1998_Bioorg.Med.Chem_6_427 |
| Author(s) : Badia A , Banos JE , Camps P , Contreras J , Gorbig DM , Munoz-Torrero D , Simon M , Vivas NM |
| Ref : Bioorganic & Medicinal Chemistry , 6 :427 , 1998 |
| Abstract : Badia_1998_Bioorg.Med.Chem_6_427 |
| ESTHER : Badia_1998_Bioorg.Med.Chem_6_427 |
| PubMedSearch : Badia_1998_Bioorg.Med.Chem_6_427 |
| PubMedID: 9597187 |
| Title : Nonequilibrium analysis alters the mechanistic interpretation of inhibition of acetylcholinesterase by peripheral site ligands - Szegletes_1998_Biochemistry_37_4206 |
| Author(s) : Szegletes T , Mallender WD , Rosenberry TL |
| Ref : Biochemistry , 37 :4206 , 1998 |
| Abstract : Szegletes_1998_Biochemistry_37_4206 |
| ESTHER : Szegletes_1998_Biochemistry_37_4206 |
| PubMedSearch : Szegletes_1998_Biochemistry_37_4206 |
| PubMedID: 9521743 |
| Title : Comparative studies of huperzine A, E2020, and tacrine on behavior and cholinesterase activities - Cheng_1998_Pharmacol.Biochem.Behav_60_377 |
| Author(s) : Cheng DH , Tang XC |
| Ref : Pharmacol Biochem Behav , 60 :377 , 1998 |
| Abstract : Cheng_1998_Pharmacol.Biochem.Behav_60_377 |
| ESTHER : Cheng_1998_Pharmacol.Biochem.Behav_60_377 |
| PubMedSearch : Cheng_1998_Pharmacol.Biochem.Behav_60_377 |
| PubMedID: 9632220 |
| Title : Inhibition of cholinesterase-associated aryl acylamidase activity by anticholinesterase agents: focus on drugs potentially effective in Alzheimer's disease - Costagli_1998_Biochem.Pharmacol_55_1733 |
| Author(s) : Costagli C , Galli A |
| Ref : Biochemical Pharmacology , 55 :1733 , 1998 |
| Abstract : Costagli_1998_Biochem.Pharmacol_55_1733 |
| ESTHER : Costagli_1998_Biochem.Pharmacol_55_1733 |
| PubMedSearch : Costagli_1998_Biochem.Pharmacol_55_1733 |
| PubMedID: 9634011 |
| Title : Reversal of scopolamine-induced deficits in radial maze performance by (-)-huperzine A: comparison with E2020 and tacrine - Wang_1998_Eur.J.Pharmacol_349_137 |
| Author(s) : Wang T , Tang XC |
| Ref : European Journal of Pharmacology , 349 :137 , 1998 |
| Abstract : Wang_1998_Eur.J.Pharmacol_349_137 |
| ESTHER : Wang_1998_Eur.J.Pharmacol_349_137 |
| PubMedSearch : Wang_1998_Eur.J.Pharmacol_349_137 |
| PubMedID: 9671090 |
| Title : Structure of acetylcholinesterase complexed with the nootropic alkaloid, (-)-huperzine A - Raves_1997_Nat.Struct.Biol_4_57 |
| Author(s) : Raves ML , Harel M , Pang YP , Silman I , Kozikowski AP , Sussman JL |
| Ref : Nat Struct Biol , 4 :57 , 1997 |
| Abstract : Raves_1997_Nat.Struct.Biol_4_57 |
| ESTHER : Raves_1997_Nat.Struct.Biol_4_57 |
| PubMedSearch : Raves_1997_Nat.Struct.Biol_4_57 |
| PubMedID: 8989325 |
| Gene_locus related to this paper: torca-ACHE |
| Title : Huperzine A, a novel promising acetylcholinesterase inhibitor - Cheng_1996_Neuroreport_8_97 |
| Author(s) : Cheng DH , Ren H , Tang XC |
| Ref : Neuroreport , 8 :97 , 1996 |
| Abstract : Cheng_1996_Neuroreport_8_97 |
| ESTHER : Cheng_1996_Neuroreport_8_97 |
| PubMedSearch : Cheng_1996_Neuroreport_8_97 |
| PubMedID: 9051760 |
| Title : Effects of isovanihuperzine A on cholinesterase and scopolamine-induced memory impairment - Xiong_1995_Zhongguo.Yao.Li.Xue.Bao_16_21 |
| Author(s) : Xiong ZQ , Tang XC , Lin JL , Zhu DY |
| Ref : Acta Pharmacol Sin , 16 :21 , 1995 |
| Abstract : Xiong_1995_Zhongguo.Yao.Li.Xue.Bao_16_21 |
| ESTHER : Xiong_1995_Zhongguo.Yao.Li.Xue.Bao_16_21 |
| PubMedSearch : Xiong_1995_Zhongguo.Yao.Li.Xue.Bao_16_21 |
| PubMedID: 7771190 |
| Title : Synthesis and evaluation of 5-amino-5,6,7,8-tetrahydroquinolinones as potential agents for the treatment of Alzheimer's disease - Fink_1995_J.Med.Chem_38_3645 |
| Author(s) : Fink DM , Bores GM , Effland RC , Huger FP , Kurys BE , Rush DK , Selk DE |
| Ref : Journal of Medicinal Chemistry , 38 :3645 , 1995 |
| Abstract : Fink_1995_J.Med.Chem_38_3645 |
| ESTHER : Fink_1995_J.Med.Chem_38_3645 |
| PubMedSearch : Fink_1995_J.Med.Chem_38_3645 |
| PubMedID: 7658452 |
| Title : Identification of amino acid residues involved in the binding of Huperzine A to cholinesterases - Saxena_1994_Protein.Sci_3_1770 |
| Author(s) : Saxena A , Qian N , Kovach IM , Kozikowski AP , Pang YP , Vellom DC , Radic Z , Quinn DM , Taylor P , Doctor BP |
| Ref : Protein Science , 3 :1770 , 1994 |
| Abstract : Saxena_1994_Protein.Sci_3_1770 |
| ESTHER : Saxena_1994_Protein.Sci_3_1770 |
| PubMedSearch : Saxena_1994_Protein.Sci_3_1770 |
| PubMedID: 7849595 |
| Title : Comparison of the effects of natural and synthetic huperzine-A on rat brain cholinergic function in vitro and in vivo - Tang_1994_J.Ethnopharmacol_44_147 |
| Author(s) : Tang XC , Kindel GH , Kozikowski AP , Hanin I |
| Ref : J Ethnopharmacol , 44 :147 , 1994 |
| Abstract : Tang_1994_J.Ethnopharmacol_44_147 |
| ESTHER : Tang_1994_J.Ethnopharmacol_44_147 |
| PubMedSearch : Tang_1994_J.Ethnopharmacol_44_147 |
| PubMedID: 7898122 |
| Title : Prediction of the binding sites of huperzine A in acetylcholinesterase by docking studies - Pang_1994_J.Comput.Aided.Mol.Des_8_669 |
| Author(s) : Pang YP , Kozikowski AP |
| Ref : J Comput Aided Mol Des , 8 :669 , 1994 |
| Abstract : Pang_1994_J.Comput.Aided.Mol.Des_8_669 |
| ESTHER : Pang_1994_J.Comput.Aided.Mol.Des_8_669 |
| PubMedSearch : Pang_1994_J.Comput.Aided.Mol.Des_8_669 |
| PubMedID: 7738603 |
| Title : Role of tyrosine 337 in the binding of huperzine A to the active site of human acetylcholinesterase - Ashani_1994_Mol.Pharmacol_45_555 |
| Author(s) : Ashani Y , Grunwald J , Kronman C , Velan B , Shafferman A |
| Ref : Molecular Pharmacology , 45 :555 , 1994 |
| Abstract : Ashani_1994_Mol.Pharmacol_45_555 |
| ESTHER : Ashani_1994_Mol.Pharmacol_45_555 |
| PubMedSearch : Ashani_1994_Mol.Pharmacol_45_555 |
| PubMedID: 8145739 |
| Gene_locus related to this paper: human-ACHE |
| Title : Natural and synthetic Huperzine A: effect on cholinergic function in vitro and in vivo - Hanin_1993_Ann.N.Y.Acad.Sci_695_304 |
| Author(s) : Hanin I , Tang XC , Kindel GL , Kozikowski AP |
| Ref : Annals of the New York Academy of Sciences , 695 :304 , 1993 |
| Abstract : Hanin_1993_Ann.N.Y.Acad.Sci_695_304 |
| ESTHER : Hanin_1993_Ann.N.Y.Acad.Sci_695_304 |
| PubMedSearch : Hanin_1993_Ann.N.Y.Acad.Sci_695_304 |
| PubMedID: 8239300 |
| Title : Mechanism of inhibition of cholinesterases by huperzine A - Ashani_1992_Biochem.Biophys.Res.Commun_184_719 |
| Author(s) : Ashani Y , Peggins JO, 3rd , Doctor BP |
| Ref : Biochemical & Biophysical Research Communications , 184 :719 , 1992 |
| Abstract : Ashani_1992_Biochem.Biophys.Res.Commun_184_719 |
| ESTHER : Ashani_1992_Biochem.Biophys.Res.Commun_184_719 |
| PubMedSearch : Ashani_1992_Biochem.Biophys.Res.Commun_184_719 |
| PubMedID: 1575745 |
| Title : Huperzine A--a potent acetylcholinesterase inhibitor of use in the treatment of Alzheimer's disease - Geib_1991_Acta.Crystallogr_C824 |
| Author(s) : Geib SJ , Tuckmantel W , Kozikowski AP |
| Ref : Acta Crystallographica , C47 pt4 :824 , 1991 |
| Abstract : Geib_1991_Acta.Crystallogr_C824 |
| ESTHER : Geib_1991_Acta.Crystallogr_C824 |
| PubMedSearch : Geib_1991_Acta.Crystallogr_C824 |
| PubMedID: 1863425 |
| Title : Development of natural products as drugs acting on central nervous system - Zhu_1991_Mem.Inst.Oswaldo.Cruz_86_173 |
| Author(s) : Zhu XZ |
| Ref : Mem Inst Oswaldo Cruz , 86 Suppl 2 :173 , 1991 |
| Abstract : Zhu_1991_Mem.Inst.Oswaldo.Cruz_86_173 |
| ESTHER : Zhu_1991_Mem.Inst.Oswaldo.Cruz_86_173 |
| PubMedSearch : Zhu_1991_Mem.Inst.Oswaldo.Cruz_86_173 |
| PubMedID: 1841995 |