Mequitazine
General
Type : Phenothiazine,Sulfur Compound,Not A\/B H target,Azabicyclo
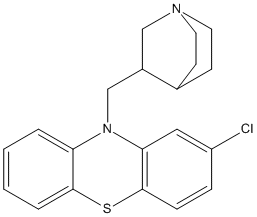
Chemical_Nomenclature : 10-(1-azabicyclo[2.2.2]octan-3-ylmethyl)phenothiazine
Canonical SMILES : C1CN2CCC1C(C2)CN3C4=CC=CC=C4SC5=CC=CC=C53
InChI : InChI=1S\/C20H22N2S\/c1-3-7-19-17(5-1)22(18-6-2-4-8-20(18)23-19)14-16-13-21-11-9-15(16)10-12-21\/h1-8,15-16H,9-14H2
InChIKey : HOKDBMAJZXIPGC-UHFFFAOYSA-N
Other name(s) : Metaplexan,Primalan,Virginan
MW : 322.5
Formula : C20H22N2S
CAS_number : 29216-28-2
PubChem : 4066
UniChem : HOKDBMAJZXIPGC-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
Families :
Stucture :
Protein :
References (4)
| Title : Inhibition of butyrylcholinesterase by phenothiazine derivatives - Debord_2002_J.Enzyme.Inhib.Med.Chem_17_197 |
| Author(s) : Debord J , Merle L , Bollinger JC , Dantoine T |
| Ref : J Enzyme Inhib Med Chem , 17 :197 , 2002 |
| Abstract : Debord_2002_J.Enzyme.Inhib.Med.Chem_17_197 |
| ESTHER : Debord_2002_J.Enzyme.Inhib.Med.Chem_17_197 |
| PubMedSearch : Debord_2002_J.Enzyme.Inhib.Med.Chem_17_197 |
| PubMedID: 12443046 |
| Title : An In Vitro Study on the Metabolism and Possible Drug Interactions of Rokitamycin, a Macrolide Antibiotic, Using Human Liver Microsomes - Zhao_1999_Drug.Metab.Dispos_27_776 |
| Author(s) : Zhao XJ , Koyama E , Ishizaki T |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 27 :776 , 1999 |
| Abstract : Zhao_1999_Drug.Metab.Dispos_27_776 |
| ESTHER : Zhao_1999_Drug.Metab.Dispos_27_776 |
| PubMedSearch : Zhao_1999_Drug.Metab.Dispos_27_776 |
| PubMedID: 10383920 |
| Title : An in-vitro study on the metabolism of rokitamycin and possible interactions of the drug with rat liver microsomes - Zhao_1999_J.Pharm.Pharmacol_51_1167 |
| Author(s) : Zhao XJ , Ishizaki T |
| Ref : J Pharm Pharmacol , 51 :1167 , 1999 |
| Abstract : Zhao_1999_J.Pharm.Pharmacol_51_1167 |
| ESTHER : Zhao_1999_J.Pharm.Pharmacol_51_1167 |
| PubMedSearch : Zhao_1999_J.Pharm.Pharmacol_51_1167 |
| PubMedID: 10579688 |
| Title : Antagonism of the five cloned human muscarinic cholinergic receptors expressed in CHO-K1 cells by antidepressants and antihistaminics - Stanton_1993_Biochem.Pharmacol_45_2352 |
| Author(s) : Stanton T , Bolden-Watson C , Cusack B , Richelson E |
| Ref : Biochemical Pharmacology , 45 :2352 , 1993 |
| Abstract : Stanton_1993_Biochem.Pharmacol_45_2352 |
| ESTHER : Stanton_1993_Biochem.Pharmacol_45_2352 |
| PubMedSearch : Stanton_1993_Biochem.Pharmacol_45_2352 |
| PubMedID: 8100134 |