Orlistat
Tetrahydrolipstatin. THL. Non-specific lipase inhibitor. Orlistat is a drug designed to treat obesity. Its primary function is preventing the absorption of fats from the human diet, thereby reducing caloric intake. Orlistat works by inhibiting pancreatic lipase, an enzyme that breaks down triglycerides in the intestine. Without this enzyme, triglycerides from the diet are prevented from being hydrolyzed into absorbable free fatty acids and are excreted undigested. Inhibits also Sn1-specific DAGLA and DAGLB. Inhibits CES2, FASN, PNLIPRP2, ABHD16A, . Orlistat is a saturated derivative of the natural compound Lipstatin, isolated from Streptomyces toxytricini
General
Type : Oxooxetan,Lipase inhibitor
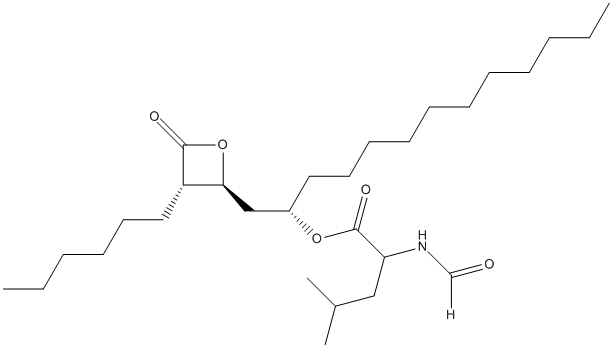
Chemical_Nomenclature : [(2S)-1-[(2S,3S)-3-hexyl-4-oxooxetan-2-yl]tridecan-2-yl] (2S)-2-formamido-4-methylpentanoate
Canonical SMILES : CCCCCCCCCCCC(CC1C(C(=O)O1)CCCCCC)OC(=O)C(CC(C)C)NC=O
InChI : InChI=1S\/C29H53NO5\/c1-5-7-9-11-12-13-14-15-16-18-24(34-29(33)26(30-22-31)20-23(3)4)21-27-25(28(32)35-27)19-17-10-8-6-2\/h22-27H,5-21H2,1-4H3,(H,30,31)\/t24-,25-,26-,27-\/m0\/s1 || InChI=1S\/C29H53NO5\/c1-5-7-9-11-12-13-14-15-16-18-24(34-29(33)26(30-22-31)20-23(3)4)21-27-25(28(32)35-27)19-17-10-8-6-2\/h22-27H,5-21H2,1-4H3,(H,30,31)
InChIKey : AHLBNYSZXLDEJQ-FWEHEUNISA-N || AHLBNYSZXLDEJQ-UHFFFAOYSA-N
Other name(s) : Tetrahydrolipstatin,Orlipastat,Xenical,Alli,(-)-Tetrahydrolipstatin,Orlipastatum,THLP,Lipase Inhibitor,THL,1-(3-hexyl-4-oxooxetan-2-yl)tridecan-2-yl 2-formamido-4-methylpentanoate,Orlistate,NCGC00095128-01,Orlistat, 5,ACMC-20p1ed
MW : 495.73
Formula : C29H53NO5
CAS_number : 96829-58-2
PubChem : 3034010
UniChem : AHLBNYSZXLDEJQ-FWEHEUNISA-N || AHLBNYSZXLDEJQ-UHFFFAOYSA-N
IUPHAR : 5277
Wikipedia : Orlistat
Target
Families : Orlistat ligand of proteins in family: AlphaBeta_hydrolase || Acidic_Lipase || Pancreatic_lipase || Thioesterase || Carb_B_Chordata || ABHD6-Lip || Lipase_3 || Hormone-sensitive_lipase_like || Monoglyceridelipase_lysophospholip || Cutinase || ABHD16 || A85-Mycolyl-transferase || PE-PPE || Plasmodium_subtelomeric_PST-A || Bacterial_lip_FamI.6
Stucture :
References (27)
| Title : Evaluation of drug-drug interactions via inhibition of hydrolases by orlistat, an anti-obesity drug - Hirosawa_2023_Drug.Metab.Dispos__ |
| Author(s) : Hirosawa K , Fukami T , Nakano M , Nakajima M |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , : , 2023 |
| Abstract : Hirosawa_2023_Drug.Metab.Dispos__ |
| ESTHER : Hirosawa_2023_Drug.Metab.Dispos__ |
| PubMedSearch : Hirosawa_2023_Drug.Metab.Dispos__ |
| PubMedID: 37137721 |
| Title : Identification of cell wall synthesis inhibitors active against Mycobacterium tuberculosis by competitive activity-based protein profiling - Li_2022_Cell.Chem.Biol_29_883 |
| Author(s) : Li M , Patel HV , Cognetta AB, 3rd , Smith TC, 2nd , Mallick I , Cavalier JF , Previti ML , Canaan S , Aldridge BB , Cravatt BF , Seeliger JC |
| Ref : Cell Chemical Biology , 29 :883 , 2022 |
| Abstract : Li_2022_Cell.Chem.Biol_29_883 |
| ESTHER : Li_2022_Cell.Chem.Biol_29_883 |
| PubMedSearch : Li_2022_Cell.Chem.Biol_29_883 |
| PubMedID: 34599873 |
| Title : Total Synthesis of Tetrahydrolipstatin, Its Derivatives, and Evaluation of Their Ability to Potentiate Multiple Antibiotic Classes against Mycobacterium Species - Khan_2021_ACS.Infect.Dis_7_2876 |
| Author(s) : Khan SS , Sudasinghe TD , Landgraf AD , Ronning DR , Sucheck SJ |
| Ref : ACS Infect Dis , 7 :2876 , 2021 |
| Abstract : Khan_2021_ACS.Infect.Dis_7_2876 |
| ESTHER : Khan_2021_ACS.Infect.Dis_7_2876 |
| PubMedSearch : Khan_2021_ACS.Infect.Dis_7_2876 |
| PubMedID: 34478259 |
| Gene_locus related to this paper: myctu-a85c |
| Title : Lysosomal acid lipase is the major acid retinyl ester hydrolase in cultured human hepatic stellate cells but not essential for retinyl ester degradation - Wagner_2020_Biochim.Biophys.Acta.Mol.Cell.Biol.Lipids_1865_158730 |
| Author(s) : Wagner C , Hois V , Pajed L , Pusch LM , Wolinski H , Trauner M , Zimmermann R , Taschler U , Lass A |
| Ref : Biochimica & Biophysica Acta Molecular & Cellular Biology Lipids , 1865 :158730 , 2020 |
| Abstract : Wagner_2020_Biochim.Biophys.Acta.Mol.Cell.Biol.Lipids_1865_158730 |
| ESTHER : Wagner_2020_Biochim.Biophys.Acta.Mol.Cell.Biol.Lipids_1865_158730 |
| PubMedSearch : Wagner_2020_Biochim.Biophys.Acta.Mol.Cell.Biol.Lipids_1865_158730 |
| PubMedID: 32361002 |
| Gene_locus related to this paper: human-LIPA |
| Title : Crystal structure of pathogenic Staphylococcus aureus lipase complex with the anti-obesity drug orlistat - Kitadokoro_2020_Sci.Rep_10_5469 |
| Author(s) : Kitadokoro K , Tanaka M , Hikima T , Okuno Y , Yamamoto M , Kamitani S |
| Ref : Sci Rep , 10 :5469 , 2020 |
| Abstract : Kitadokoro_2020_Sci.Rep_10_5469 |
| ESTHER : Kitadokoro_2020_Sci.Rep_10_5469 |
| PubMedSearch : Kitadokoro_2020_Sci.Rep_10_5469 |
| PubMedID: 32214208 |
| Gene_locus related to this paper: staau-lipas |
| Title : The Antimalarial Natural Product Salinipostin A Identifies Essential alpha\/beta Serine Hydrolases Involved in Lipid Metabolism in P. falciparum Parasites - Yoo_2020_Cell.Chem.Biol_27_143 |
| Author(s) : Yoo E , Schulze CJ , Stokes BH , Onguka O , Yeo T , Mok S , Gnadig NF , Zhou Y , Kurita K , Foe IT , Terrell SM , Boucher MJ , Cieplak P , Kumpornsin K , Lee MCS , Linington RG , Long JZ , Uhlemann AC , Weerapana E , Fidock DA , Bogyo M |
| Ref : Cell Chemical Biology , 27 :143 , 2020 |
| Abstract : Yoo_2020_Cell.Chem.Biol_27_143 |
| ESTHER : Yoo_2020_Cell.Chem.Biol_27_143 |
| PubMedSearch : Yoo_2020_Cell.Chem.Biol_27_143 |
| PubMedID: 31978322 |
| Gene_locus related to this paper: plaf7-q8ii19 , plafa-a0a143zya4 , plaf7-q8iik5 , plafa-MAL8P1.38 , plafa-PF07.0040 , plafa-PF10.0020 , plafa-PF10.0379 , plafa-PF13.0153 |
| Title : Human leukocytes differentially express endocannabinoid-glycerol lipases and hydrolyze 2-arachidonoyl-glycerol and its metabolites from the 15-lipoxygenase and cyclooxygenase pathways - Turcotte_2019_J.Leukoc.Biol_106_1337 |
| Author(s) : Turcotte C , Dumais E , Archambault AS , Martin C , Blanchet MR , Bissonnette E , Boulet LP , Laviolette M , Di Marzo V , Flamand N |
| Ref : J Leukoc Biol , 106 :1337 , 2019 |
| Abstract : Turcotte_2019_J.Leukoc.Biol_106_1337 |
| ESTHER : Turcotte_2019_J.Leukoc.Biol_106_1337 |
| PubMedSearch : Turcotte_2019_J.Leukoc.Biol_106_1337 |
| PubMedID: 31556464 |
| Gene_locus related to this paper: human-ABHD6 , human-ABHD12 , human-ABHD16A , human-CES1 , human-LYPLA2 , human-PPT1 |
| Title : Mycolyltransferase from Mycobacterium tuberculosis in covalent complex with tetrahydrolipstatin provides insights into antigen 85 catalysis - Goins_2018_J.Biol.Chem_293_3651 |
| Author(s) : Goins CM , Dajnowicz S , Smith MD , Parks JM , Ronning DR |
| Ref : Journal of Biological Chemistry , 293 :3651 , 2018 |
| Abstract : Goins_2018_J.Biol.Chem_293_3651 |
| ESTHER : Goins_2018_J.Biol.Chem_293_3651 |
| PubMedSearch : Goins_2018_J.Biol.Chem_293_3651 |
| PubMedID: 29352107 |
| Gene_locus related to this paper: myctu-a85c |
| Title : Characterization of Tetrahydrolipstatin and Stereoderivatives on the Inhibition of Essential Mycobacterium tuberculosis Lipid Esterases - Goins_2018_Biochemistry_57_2383 |
| Author(s) : Goins CM , Sudasinghe TD , Liu X , Wang Y , O'Doherty GA , Ronning DR |
| Ref : Biochemistry , 57 :2383 , 2018 |
| Abstract : Goins_2018_Biochemistry_57_2383 |
| ESTHER : Goins_2018_Biochemistry_57_2383 |
| PubMedSearch : Goins_2018_Biochemistry_57_2383 |
| PubMedID: 29601187 |
| Gene_locus related to this paper: myctu-a85c , myctu-PKS13 , myctu-Rv3802c |
| Title : Stereochemical Structure Activity Relationship Studies (S-SAR) of Tetrahydrolipstatin - Liu_2018_ACS.Med.Chem.Lett_9_274 |
| Author(s) : Liu X , Wang Y , Duclos RI, Jr. , O'Doherty GA |
| Ref : ACS Med Chem Lett , 9 :274 , 2018 |
| Abstract : Liu_2018_ACS.Med.Chem.Lett_9_274 |
| ESTHER : Liu_2018_ACS.Med.Chem.Lett_9_274 |
| PubMedSearch : Liu_2018_ACS.Med.Chem.Lett_9_274 |
| PubMedID: 29541373 |
| Title : Lipase inhibitor orlistat prevents hepatitis B virus infection by targeting an early step in the virus life cycle - Esser_2018_Antiviral.Res_151_4 |
| Author(s) : Esser K , Lucifora J , Wettengel J , Singethan K , Glinzer A , Zernecke A , Protzer U |
| Ref : Antiviral Res , 151 :4 , 2018 |
| Abstract : Esser_2018_Antiviral.Res_151_4 |
| ESTHER : Esser_2018_Antiviral.Res_151_4 |
| PubMedSearch : Esser_2018_Antiviral.Res_151_4 |
| PubMedID: 29309795 |
| Title : Simplified assays of lipolysis enzymes for drug discovery and specificity assessment of known inhibitors - Iglesias_2016_J.Lipid.Res_57_131 |
| Author(s) : Iglesias J , Lamontagne J , Erb H , Gezzar S , Zhao S , Joly E , Truong VL , Skorey K , Crane S , Madiraju SR , Prentki M |
| Ref : J Lipid Res , 57 :131 , 2016 |
| Abstract : Iglesias_2016_J.Lipid.Res_57_131 |
| ESTHER : Iglesias_2016_J.Lipid.Res_57_131 |
| PubMedSearch : Iglesias_2016_J.Lipid.Res_57_131 |
| PubMedID: 26423520 |
| Title : Targeting Lipid Esterases in Mycobacteria Grown Under Different Physiological Conditions Using Activity-based Profiling with Tetrahydrolipstatin (THL) - Ravindran_2014_Mol.Cell.Proteomics_13_435 |
| Author(s) : Ravindran MS , Rao SP , Cheng X , Shukla A , Cazenave-Gassiot A , Yao SQ , Wenk MR |
| Ref : Mol Cell Proteomics , 13 :435 , 2014 |
| Abstract : Ravindran_2014_Mol.Cell.Proteomics_13_435 |
| ESTHER : Ravindran_2014_Mol.Cell.Proteomics_13_435 |
| PubMedSearch : Ravindran_2014_Mol.Cell.Proteomics_13_435 |
| PubMedID: 24345785 |
| Gene_locus related to this paper: myctu-Rv2970c |
| Title : Biochemical and Pharmacological Characterization of the Human Lymphocyte Antigen B-Associated Transcript 5 (BAT5\/ABHD16A) - Savinainen_2014_PLoS.One_9_e109869 |
| Author(s) : Savinainen JR , Patel JZ , Parkkari T , Navia-Paldanius D , Marjamaa JJ , Laitinen T , Nevalainen T , Laitinen JT |
| Ref : PLoS ONE , 9 :e109869 , 2014 |
| Abstract : Savinainen_2014_PLoS.One_9_e109869 |
| ESTHER : Savinainen_2014_PLoS.One_9_e109869 |
| PubMedSearch : Savinainen_2014_PLoS.One_9_e109869 |
| PubMedID: 25290914 |
| Gene_locus related to this paper: human-ABHD16A , mouse-Abhd16a |
| Title : Carboxylesterase-2 is a highly sensitive target of the antiobesity agent orlistat with profound implications in the activation of anticancer prodrugs - Xiao_2013_Biochem.Pharmacol_85_439 |
| Author(s) : Xiao D , Shi D , Yang D , Barthel B , Koch TH , Yan B |
| Ref : Biochemical Pharmacology , 85 :439 , 2013 |
| Abstract : Xiao_2013_Biochem.Pharmacol_85_439 |
| ESTHER : Xiao_2013_Biochem.Pharmacol_85_439 |
| PubMedSearch : Xiao_2013_Biochem.Pharmacol_85_439 |
| PubMedID: 23228697 |
| Gene_locus related to this paper: human-CES2 |
| Title : Elucidation and chemical modulation of sulfolipid-1 biosynthesis in Mycobacterium tuberculosis - Seeliger_2012_J.Biol.Chem_287_7990 |
| Author(s) : Seeliger JC , Holsclaw CM , Schelle MW , Botyanszki Z , Gilmore SA , Tully SE , Niederweis M , Cravatt BF , Leary JA , Bertozzi CR |
| Ref : Journal of Biological Chemistry , 287 :7990 , 2012 |
| Abstract : Seeliger_2012_J.Biol.Chem_287_7990 |
| ESTHER : Seeliger_2012_J.Biol.Chem_287_7990 |
| PubMedSearch : Seeliger_2012_J.Biol.Chem_287_7990 |
| PubMedID: 22194604 |
| Gene_locus related to this paper: myctu-Rv3822 |
| Title : Tetrahydrolipstatin inhibition, functional analyses, and three-dimensional structure of a lipase essential for mycobacterial viability - Crellin_2010_J.Biol.Chem_285_30050 |
| Author(s) : Crellin PK , Vivian JP , Scoble J , Chow FM , West NP , Brammananth R , Proellocks NI , Shahine A , Le Nours J , Wilce MC , Britton WJ , Coppel RL , Rossjohn J , Beddoe T |
| Ref : Journal of Biological Chemistry , 285 :30050 , 2010 |
| Abstract : Crellin_2010_J.Biol.Chem_285_30050 |
| ESTHER : Crellin_2010_J.Biol.Chem_285_30050 |
| PubMedSearch : Crellin_2010_J.Biol.Chem_285_30050 |
| PubMedID: 20656688 |
| Gene_locus related to this paper: mycs2-a0r619 |
| Title : Trp-107 and trp-253 account for the increased steady state fluorescence that accompanies the conformational change in human pancreatic triglyceride lipase induced by tetrahydrolipstatin and bile salt - Bourbon-Freie_2009_J.Biol.Chem_284_14157 |
| Author(s) : Bourbon-Freie A , Dub RE , Xiao X , Lowe ME |
| Ref : Journal of Biological Chemistry , 284 :14157 , 2009 |
| Abstract : Bourbon-Freie_2009_J.Biol.Chem_284_14157 |
| ESTHER : Bourbon-Freie_2009_J.Biol.Chem_284_14157 |
| PubMedSearch : Bourbon-Freie_2009_J.Biol.Chem_284_14157 |
| PubMedID: 19346257 |
| Gene_locus related to this paper: human-PNLIP |
| Title : Asymmetric synthesis of anti-aldol segments via a nonaldol route: synthetic applications to statines and (-)-tetrahydrolipstatin - Ghosh_2009_J.Org.Chem_74_4508 |
| Author(s) : Ghosh AK , Shurrush K , Kulkarni S |
| Ref : J Org Chem , 74 :4508 , 2009 |
| Abstract : Ghosh_2009_J.Org.Chem_74_4508 |
| ESTHER : Ghosh_2009_J.Org.Chem_74_4508 |
| PubMedSearch : Ghosh_2009_J.Org.Chem_74_4508 |
| PubMedID: 19438217 |
| Title : Tetrahydrolipstatin analogues as modulators of endocannabinoid 2-arachidonoylglycerol metabolism - Ortar_2008_J.Med.Chem_51_6970 |
| Author(s) : Ortar G , Bisogno T , Ligresti A , Morera E , Nalli M , Di Marzo V |
| Ref : Journal of Medicinal Chemistry , 51 :6970 , 2008 |
| Abstract : Ortar_2008_J.Med.Chem_51_6970 |
| ESTHER : Ortar_2008_J.Med.Chem_51_6970 |
| PubMedSearch : Ortar_2008_J.Med.Chem_51_6970 |
| PubMedID: 18831576 |
| Title : Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat - Pemble_2007_Nat.Struct.Mol.Biol_14_704 |
| Author(s) : Pemble CWt , Johnson LC , Kridel SJ , Lowther WT |
| Ref : Nat Struct Mol Biol , 14 :704 , 2007 |
| Abstract : Pemble_2007_Nat.Struct.Mol.Biol_14_704 |
| ESTHER : Pemble_2007_Nat.Struct.Mol.Biol_14_704 |
| PubMedSearch : Pemble_2007_Nat.Struct.Mol.Biol_14_704 |
| PubMedID: 17618296 |
| Gene_locus related to this paper: human-FASN |
| Title : Further biochemical characterization of human pancreatic lipase-related protein 2 expressed in yeast cells - Eydoux_2007_J.Lipid.Res_48_1539 |
| Author(s) : Eydoux C , De Caro J , Ferrato F , Boullanger P , Lafont D , Laugier R , Carriere F , de Caro A |
| Ref : J Lipid Res , 48 :1539 , 2007 |
| Abstract : Eydoux_2007_J.Lipid.Res_48_1539 |
| ESTHER : Eydoux_2007_J.Lipid.Res_48_1539 |
| PubMedSearch : Eydoux_2007_J.Lipid.Res_48_1539 |
| PubMedID: 17401110 |
| Gene_locus related to this paper: human-PNLIPRP2 |
| Title : Mass spectrometric evidence of covalently-bound tetrahydrolipstatin at the catalytic serine of Streptomyces rimosus lipase - Asler_2007_Biochim.Biophys.Acta_1770_163 |
| Author(s) : Asler IL , Zehl M , Kovacic F , Muller R , Abramic M , Allmaier G , Kojic-Prodic B |
| Ref : Biochimica & Biophysica Acta , 1770 :163 , 2007 |
| Abstract : Asler_2007_Biochim.Biophys.Acta_1770_163 |
| ESTHER : Asler_2007_Biochim.Biophys.Acta_1770_163 |
| PubMedSearch : Asler_2007_Biochim.Biophys.Acta_1770_163 |
| PubMedID: 17137716 |
| Title : Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity - Kridel_2004_Cancer.Res_64_2070 |
| Author(s) : Kridel SJ , Axelrod F , Rozenkrantz N , Smith JW |
| Ref : Cancer Research , 64 :2070 , 2004 |
| Abstract : Kridel_2004_Cancer.Res_64_2070 |
| ESTHER : Kridel_2004_Cancer.Res_64_2070 |
| PubMedSearch : Kridel_2004_Cancer.Res_64_2070 |
| PubMedID: 15026345 |
| Gene_locus related to this paper: human-FASN |
| Title : Inhibition of gastrointestinal lipolysis by Orlistat during digestion of test meals in healthy volunteers - Carriere_2001_Am.J.Physiol.Gastrointest.Liver.Physiol_281_G16 |
| Author(s) : Carriere F , Renou C , Ransac S , Lopez V , De Caro J , Ferrato F , de Caro A , Fleury A , Sanwald-Ducray P , Lengsfeld H , Beglinger C , Hadvary P , Verger R , Laugier R |
| Ref : American Journal of Physiology Gastrointest Liver Physiol , 281 :G16 , 2001 |
| Abstract : Carriere_2001_Am.J.Physiol.Gastrointest.Liver.Physiol_281_G16 |
| ESTHER : Carriere_2001_Am.J.Physiol.Gastrointest.Liver.Physiol_281_G16 |
| PubMedSearch : Carriere_2001_Am.J.Physiol.Gastrointest.Liver.Physiol_281_G16 |
| PubMedID: 11408251 |
| Title : Orlistat F Hoffmann-La Roche Ltd - Malone_1998_IDrugs_1_232 |
| Author(s) : Malone M |
| Ref : IDrugs , 1 :232 , 1998 |
| Abstract : Malone_1998_IDrugs_1_232 |
| ESTHER : Malone_1998_IDrugs_1_232 |
| PubMedSearch : Malone_1998_IDrugs_1_232 |
| PubMedID: 18465537 |
| Title : Mode of action of orlistat - Guerciolini_1997_Int.J.Obes.Relat.Metab.Disord_21 Suppl 3_S12 |
| Author(s) : Guerciolini R |
| Ref : Int J Obes Relat Metab Disord , 21 Suppl 3 :S12 , 1997 |
| Abstract : Guerciolini_1997_Int.J.Obes.Relat.Metab.Disord_21 Suppl 3_S12 |
| ESTHER : Guerciolini_1997_Int.J.Obes.Relat.Metab.Disord_21 Suppl 3_S12 |
| PubMedSearch : Guerciolini_1997_Int.J.Obes.Relat.Metab.Disord_21 Suppl 3_S12 |
| PubMedID: 9225172 |