Physostigmine~Eserine
Carbamate alkaloid purified from the calabar bean: Physostigma venenosum. Physostigmine is a cholinesterase inhibitor that is rapidly absorbed through membranes. It can be applied topically to the conjunctiva. It also can cross the blood-brain barrier and is used when central nervous system effects are desired, as in the treatment of severe anticholinergic toxicity.
General
Type : Derivative of physostigmine-eserine,Natural,Carbamate,Drug,Indole
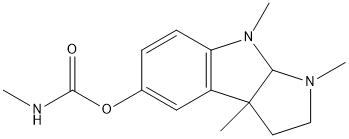
Chemical_Nomenclature : 1'methylpyrrolidino (2':3':2:3) 1,3-dimethylindolin-\%-yl N-methylcarbamate
Canonical SMILES : CC12CCN(C1N(C3=C2C=C(C=C3)OC(=O)NC)C)C || CC12CC[NH+](C1[NH+](C3=C2C=C(C=C3)OC(=O)NC)C)C.C1=CC=C(C(=C1)C(=O)[O-])[O-] || CC12CC[NH+](C1N(C3=C2C=C(C=C3)OC(=O)NC)C)C.CC12CC[NH+](C1N(C3=C2C=C(C=C3)OC(=O)NC)C)C.[O-]S(=O)(=O)[O-] || CC12CCN(C1N(C3=C2C=C(C=C3)OC(=O)NC)C)C.Br
InChI : InChI=1S\/C15H21N3O2\/c1-15-7-8-17(3)13(15)18(4)12-6-5-10(9-11(12)15)20-14(19)16-2\/h5-6,9,13H,7-8H2,1-4H3,(H,16,19) || InChI=1S\/C15H21N3O2\/c1-15-7-8-17(3)13(15)18(4)12-6-5-10(9-11(12)15)20-14(19)16-2\/h5-6,9,13H,7-8H2,1-4H3,(H,16,19)\/t13-,15+\/m1\/s1 || InChI=1S\/C15H21N3O2.C7H6O3\/c1-15-7-8-17(3)13(15)18(4)12-6-5-10(9-11(12)15)20-14(19)16-2\;8-6-4-2-1-3-5(6)7(9)10\/h5-6,9,13H,7-8H2,1-4H3,(H,16,19)\;1-4,8H,(H,9,10)\/t13-,15+\;\/m1.\/s1 || InChI=1S\/2C15H21N3O2.H2O4S\/c2*1-15-7-8-17(3)13(15)18(4)12-6-5-10(9-11(12)15)20-14(19)16-2\;1-5(2,3)4\/h2*5-6,9,13H,7-8H2,1-4H3,(H,16,19)\;(H2,1,2,3,4)\/t2*13-,15+\;\/m11.\/s1 || InChI=1S\/C15H21N3O2.BrH\/c1-15-7-8-17(3)13(15)18(4)12-6-5-10(9-11(12)15)20-14(19)16-2\;\/h5-6,9,13H,7-8H2,1-4H3,(H,16,19)\;1H\/t13-,15+\;\/m1.\/s1
InChIKey : PIJVFDBKTWXHHD-UHFFFAOYSA-N || PIJVFDBKTWXHHD-HIFRSBDPSA-N || HZOTZTANVBDFOF-PBCQUBLHSA-N || YYBNDIVPHIWTPK-KYJQVDHRSA-N || HFIWKRLOGGSAQX-PBCQUBLHSA-N
Other name(s) : Physostigmine,Antilirium,Esromiotin,Physostol,Ezerin,Calabarine,Erserine,Cogmine Erserine,Eserine sulfate,Eserine sulphate,Physostigmine SO4,Physostigmine sulphate,Physostigmine hemisulfate,Physostigmine hydrobromide,Physostigmine monohydrobromide,Isopto Eserine,Eserine salicylate,Isopto P-ES,Physostol salicylate,Physostigmine salicylate,Physostigmine monosalicylate,CCRIS 3405,Physostigmine, salicylate (1:1),EINECS 228-032-2,6091-13-0,Physotol,Anticholium,Eserine,E-serine
Target
Families : Physostigmine~Eserine ligand of proteins in family: ACHE || BCHE || Arylacetamide_deacetylase
Stucture :
Protein :
References (92)
| Title : In vitro kinetic interactions of pyridostigmine, physostigmine and soman with erythrocyte and muscle acetylcholinesterase from different species - Herkert_2011_Toxicol.Lett_206_41 |
| Author(s) : Herkert NM , Thiermann H , Worek F |
| Ref : Toxicol Lett , 206 :41 , 2011 |
| Abstract : Herkert_2011_Toxicol.Lett_206_41 |
| ESTHER : Herkert_2011_Toxicol.Lett_206_41 |
| PubMedSearch : Herkert_2011_Toxicol.Lett_206_41 |
| PubMedID: 21414391 |
| Title : Arylacetamide deacetylase is a determinant enzyme for the difference in hydrolase activities of phenacetin and acetaminophen - Watanabe_2010_Drug.Metab.Dispos_38_1532 |
| Author(s) : Watanabe A , Fukami T , Takahashi S , Kobayashi Y , Nakagawa N , Nakajima M , Yokoi T |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 38 :1532 , 2010 |
| Abstract : Watanabe_2010_Drug.Metab.Dispos_38_1532 |
| ESTHER : Watanabe_2010_Drug.Metab.Dispos_38_1532 |
| PubMedSearch : Watanabe_2010_Drug.Metab.Dispos_38_1532 |
| PubMedID: 20542992 |
| Title : beta-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies - Bartolini_2003_Biochem.Pharmacol_65_407 |
| Author(s) : Bartolini M , Bertucci C , Cavrini V , Andrisano V |
| Ref : Biochemical Pharmacology , 65 :407 , 2003 |
| Abstract : Bartolini_2003_Biochem.Pharmacol_65_407 |
| ESTHER : Bartolini_2003_Biochem.Pharmacol_65_407 |
| PubMedSearch : Bartolini_2003_Biochem.Pharmacol_65_407 |
| PubMedID: 12527333 |
| Title : Invited review: Cholinesterase inhibitors for Alzheimer's disease therapy: from tacrine to future applications - Giacobini_1998_Neurochem.Int_32_413 |
| Author(s) : Giacobini E |
| Ref : Neurochem Int , 32 :413 , 1998 |
| Abstract : Giacobini_1998_Neurochem.Int_32_413 |
| ESTHER : Giacobini_1998_Neurochem.Int_32_413 |
| PubMedSearch : Giacobini_1998_Neurochem.Int_32_413 |
| PubMedID: 9676739 |
| Title : Subchronic physostigmine pretreatment in guinea pigs: effective against soman and without side effects - Philippens_1998_Pharmacol.Biochem.Behav_59_1061 |
| Author(s) : Philippens IH , Busker RW , Wolthuis OL , Olivier B , Bruijnzeel PL , Melchers BP |
| Ref : Pharmacol Biochem Behav , 59 :1061 , 1998 |
| Abstract : Philippens_1998_Pharmacol.Biochem.Behav_59_1061 |
| ESTHER : Philippens_1998_Pharmacol.Biochem.Behav_59_1061 |
| PubMedSearch : Philippens_1998_Pharmacol.Biochem.Behav_59_1061 |
| PubMedID: 9586868 |
| Title : Effects of various drugs including organophosphorus compounds (OPC) and therapeutic compounds against OPC on DRL responding - Bizot_1998_Pharmacol.Biochem.Behav_59_1069 |
| Author(s) : Bizot JC |
| Ref : Pharmacol Biochem Behav , 59 :1069 , 1998 |
| Abstract : Bizot_1998_Pharmacol.Biochem.Behav_59_1069 |
| ESTHER : Bizot_1998_Pharmacol.Biochem.Behav_59_1069 |
| PubMedSearch : Bizot_1998_Pharmacol.Biochem.Behav_59_1069 |
| PubMedID: 9586869 |
| Title : Inhibition of cholinesterase-associated aryl acylamidase activity by anticholinesterase agents: focus on drugs potentially effective in Alzheimer's disease - Costagli_1998_Biochem.Pharmacol_55_1733 |
| Author(s) : Costagli C , Galli A |
| Ref : Biochemical Pharmacology , 55 :1733 , 1998 |
| Abstract : Costagli_1998_Biochem.Pharmacol_55_1733 |
| ESTHER : Costagli_1998_Biochem.Pharmacol_55_1733 |
| PubMedSearch : Costagli_1998_Biochem.Pharmacol_55_1733 |
| PubMedID: 9634011 |
| Title : Effects of hexamethonium, phenothiazines, propranolol and ephedrine on acetylcholinesterase carbamylation by physostigmine, aldicarb and carbaryl: interaction between the active site and the functionally distinct peripheral sites in acetylcholinesterase - Singh_1998_Comp.Biochem.Physiol.C.Pharmacol.Toxicol.Endocrinol_119_97 |
| Author(s) : Singh AK , Spassova D |
| Ref : Comparative Biochemistry & Physiology C Pharmacology Toxicology & Endocrinology , 119 :97 , 1998 |
| Abstract : Singh_1998_Comp.Biochem.Physiol.C.Pharmacol.Toxicol.Endocrinol_119_97 |
| ESTHER : Singh_1998_Comp.Biochem.Physiol.C.Pharmacol.Toxicol.Endocrinol_119_97 |
| PubMedSearch : Singh_1998_Comp.Biochem.Physiol.C.Pharmacol.Toxicol.Endocrinol_119_97 |
| PubMedID: 9568379 |
| Title : Functional activation of cerebral blood flow abolished by scopolamine is reversed by cognitive enhancers associated with cholinesterase inhibition: a positron emission tomography study in unanesthetized monkeys - Tsukada_1997_J.Pharmacol.Exp.Ther_281_1408 |
| Author(s) : Tsukada H , Kakiuchi T , Ando I , Ouchi Y |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 281 :1408 , 1997 |
| Abstract : Tsukada_1997_J.Pharmacol.Exp.Ther_281_1408 |
| ESTHER : Tsukada_1997_J.Pharmacol.Exp.Ther_281_1408 |
| PubMedSearch : Tsukada_1997_J.Pharmacol.Exp.Ther_281_1408 |
| PubMedID: 9190877 |
| Title : Total syntheses and anticholinesterase activities of (3aS)-N(8)- norphysostigmine, (3aS)-N(8)-norphenserine, their antipodal isomers, and other N(8)-substituted analogues - Yu_1997_J.Med.Chem_40_2895 |
| Author(s) : Yu QS , Pei XF , Holloway HW , Greig NH , Brossi A |
| Ref : Journal of Medicinal Chemistry , 40 :2895 , 1997 |
| Abstract : Yu_1997_J.Med.Chem_40_2895 |
| ESTHER : Yu_1997_J.Med.Chem_40_2895 |
| PubMedSearch : Yu_1997_J.Med.Chem_40_2895 |
| PubMedID: 9288171 |
| Title : Latanoprost and physostigmine have mostly additive ocular hypotensive effects in human eyes [see comments] - Linden_1997_Arch.Ophthalmol_115_857 |
| Author(s) : Linden C , Alm A |
| Ref : Archives of Ophthalmology , 115 :857 , 1997 |
| Abstract : Linden_1997_Arch.Ophthalmol_115_857 |
| ESTHER : Linden_1997_Arch.Ophthalmol_115_857 |
| PubMedSearch : Linden_1997_Arch.Ophthalmol_115_857 |
| PubMedID: 9230825 |
| Title : Central injection of physostigmine attenuates exercise-induced pressor response in conscious cats - Ally_1997_Am.J.Physiol_273_R393 |
| Author(s) : Ally A , Hand GA , Mitchell JH |
| Ref : American Journal of Physiology , 273 :R393 , 1997 |
| Abstract : Ally_1997_Am.J.Physiol_273_R393 |
| ESTHER : Ally_1997_Am.J.Physiol_273_R393 |
| PubMedSearch : Ally_1997_Am.J.Physiol_273_R393 |
| PubMedID: 9249577 |
| Title : Combined cholinergic and 5-HT2 receptor activation suppresses thalamocortical oscillations in aged rats - Jakala_1997_Pharmacol.Biochem.Behav_56_713 |
| Author(s) : Jakala P , Riekkinen P, Jr. |
| Ref : Pharmacol Biochem Behav , 56 :713 , 1997 |
| Abstract : Jakala_1997_Pharmacol.Biochem.Behav_56_713 |
| ESTHER : Jakala_1997_Pharmacol.Biochem.Behav_56_713 |
| PubMedSearch : Jakala_1997_Pharmacol.Biochem.Behav_56_713 |
| PubMedID: 9130298 |
| Title : Systemic physostigmine increases the antinociceptive effect of spinal morphine - Beilin_1997_Pain_70_217 |
| Author(s) : Beilin B , Nemirovsky AY , Zeidel A , Maibord E , Zelman V , Katz RL |
| Ref : Pain , 70 :217 , 1997 |
| Abstract : Beilin_1997_Pain_70_217 |
| ESTHER : Beilin_1997_Pain_70_217 |
| PubMedSearch : Beilin_1997_Pain_70_217 |
| PubMedID: 9150296 |
| Title : Cholinergic stimulation alters performance and task-specific regional cerebral blood flow during working memory - Furey_1997_Proc.Natl.Acad.Sci.U.S.A_94_6512 |
| Author(s) : Furey ML , Pietrini P , Haxby JV , Alexander GE , Lee HC , VanMeter J , Grady CL , Shetty U , Rapoport SI , Schapiro MB , Freo U |
| Ref : Proceedings of the National Academy of Sciences of the United States of America , 94 :6512 , 1997 |
| Abstract : Furey_1997_Proc.Natl.Acad.Sci.U.S.A_94_6512 |
| ESTHER : Furey_1997_Proc.Natl.Acad.Sci.U.S.A_94_6512 |
| PubMedSearch : Furey_1997_Proc.Natl.Acad.Sci.U.S.A_94_6512 |
| PubMedID: 9177249 |
| Title : Prejunctional regulation by endogenous and exogenous acetylcholine of adrenergic nerve function in isolated canine mesenteric arteries - Zhang_1997_Hypertens.Res_20_119 |
| Author(s) : Zhang JX , Okamura T , Toda N |
| Ref : Hypertens Res , 20 :119 , 1997 |
| Abstract : Zhang_1997_Hypertens.Res_20_119 |
| ESTHER : Zhang_1997_Hypertens.Res_20_119 |
| PubMedSearch : Zhang_1997_Hypertens.Res_20_119 |
| PubMedID: 9220276 |
| Title : Effects of cholinesterase inhibitors on the secretion of beta-amyloid precursor protein in cell cultures - Lahiri_1997_Ann.N.Y.Acad.Sci_826_416 |
| Author(s) : Lahiri DK , Farlow MR , Nurnberger JI, Jr. , Greig NH |
| Ref : Annals of the New York Academy of Sciences , 826 :416 , 1997 |
| Abstract : Lahiri_1997_Ann.N.Y.Acad.Sci_826_416 |
| ESTHER : Lahiri_1997_Ann.N.Y.Acad.Sci_826_416 |
| PubMedSearch : Lahiri_1997_Ann.N.Y.Acad.Sci_826_416 |
| PubMedID: 9329715 |
| Title : Anticholinesterase drugs stimulate phosphatidylinositol response in rat tracheal slices - Shibata_1996_Anesth.Analg_82_1211 |
| Author(s) : Shibata O , Kanairo M , Zhang S , Hasuo H , Morooka H , Fujie T , Sumikawa K |
| Ref : Anesthesia & Analgesia , 82 :1211 , 1996 |
| Abstract : Shibata_1996_Anesth.Analg_82_1211 |
| ESTHER : Shibata_1996_Anesth.Analg_82_1211 |
| PubMedSearch : Shibata_1996_Anesth.Analg_82_1211 |
| PubMedID: 8638793 |
| Title : Allosteric regulation of the binding of [3H]acetylcholine to m2 muscarinic receptors - Gnagey_1996_Biochem.Pharmacol_52_1767 |
| Author(s) : Gnagey AL , Ellis J |
| Ref : Biochemical Pharmacology , 52 :1767 , 1996 |
| Abstract : Gnagey_1996_Biochem.Pharmacol_52_1767 |
| ESTHER : Gnagey_1996_Biochem.Pharmacol_52_1767 |
| PubMedSearch : Gnagey_1996_Biochem.Pharmacol_52_1767 |
| PubMedID: 8986140 |
| Title : Acetylcholinesterase in Dendrobaena veneta (Oligochaeta: Opisthopora) is present with forms sensitive and insensitive to phosphatidylinositol phospholipase C. Biochemical characterization and histochemical localization in the nervous system - Talesa_1996_Eur.J.Biochem_238_538 |
| Author(s) : Talesa V , Romani R , Rosi G , Giovannini E |
| Ref : European Journal of Biochemistry , 238 :538 , 1996 |
| Abstract : Talesa_1996_Eur.J.Biochem_238_538 |
| ESTHER : Talesa_1996_Eur.J.Biochem_238_538 |
| PubMedSearch : Talesa_1996_Eur.J.Biochem_238_538 |
| PubMedID: 8681969 |
| Title : Histochemical demonstration of acetylcholinesterase activity in human Meibomian glands - Perra_1996_Eur.J.Histochem_40_39 |
| Author(s) : Perra MT , Serra A , Sirigu P , Turno F |
| Ref : European Journal of Histochemistry , 40 :39 , 1996 |
| Abstract : Perra_1996_Eur.J.Histochem_40_39 |
| ESTHER : Perra_1996_Eur.J.Histochem_40_39 |
| PubMedSearch : Perra_1996_Eur.J.Histochem_40_39 |
| PubMedID: 8741098 |
| Title : Solubilization, molecular forms, purification and substrate specificity of two acetylcholinesterases in the medicinal leech (Hirudo medicinalis) - Talesa_1995_Biochem.J_306_687 |
| Author(s) : Talesa V , Grauso M , Giovannini E , Rosi G , Toutant JP |
| Ref : Biochemical Journal , 306 :687 , 1995 |
| Abstract : Talesa_1995_Biochem.J_306_687 |
| ESTHER : Talesa_1995_Biochem.J_306_687 |
| PubMedSearch : Talesa_1995_Biochem.J_306_687 |
| PubMedID: 7702560 |
| Title : Interaction of chlorphenvinphos with cholinergic receptors in the rabbit hypothalamus - Gralewicz_1995_Neurotoxicol.Teratol_17_289 |
| Author(s) : Gralewicz S , Tomas T , Socko R |
| Ref : Neurotoxicology & Teratology , 17 :289 , 1995 |
| Abstract : Gralewicz_1995_Neurotoxicol.Teratol_17_289 |
| ESTHER : Gralewicz_1995_Neurotoxicol.Teratol_17_289 |
| PubMedSearch : Gralewicz_1995_Neurotoxicol.Teratol_17_289 |
| PubMedID: 7623739 |
| Title : Cholinesterase inhibitors proposed for treating dementia in Alzheimer's disease: selectivity toward human brain acetylcholinesterase compared with butyrylcholinesterase - Pacheco_1995_J.Pharmacol.Exp.Ther_274_767 |
| Author(s) : Pacheco G , Palacios-Esquivel R , Moss DE |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 274 :767 , 1995 |
| Abstract : Pacheco_1995_J.Pharmacol.Exp.Ther_274_767 |
| ESTHER : Pacheco_1995_J.Pharmacol.Exp.Ther_274_767 |
| PubMedSearch : Pacheco_1995_J.Pharmacol.Exp.Ther_274_767 |
| PubMedID: 7636741 |
| Title : Effect of physostigmine on cholinesterase activity in different parts of rat brain - Osman_1995_Arzneimittelforschung_45_663 |
| Author(s) : Osman MY , Abdel Tawab SM , Sharaf IA |
| Ref : Arzneimittelforschung , 45 :663 , 1995 |
| Abstract : Osman_1995_Arzneimittelforschung_45_663 |
| ESTHER : Osman_1995_Arzneimittelforschung_45_663 |
| PubMedSearch : Osman_1995_Arzneimittelforschung_45_663 |
| PubMedID: 7646566 |
| Title : Oxime effects on the rate constants of carbamylation and decarbamylation of acetylcholinesterase for pyridostigmine, physostigmine and insecticidal carbamates - Dawson_1995_Neurochem.Int_26_643 |
| Author(s) : Dawson RM |
| Ref : Neurochem Int , 26 :643 , 1995 |
| Abstract : Dawson_1995_Neurochem.Int_26_643 |
| ESTHER : Dawson_1995_Neurochem.Int_26_643 |
| PubMedSearch : Dawson_1995_Neurochem.Int_26_643 |
| PubMedID: 7670367 |
| Title : Acetylcholinesterase in tentacles of Octopus vulgaris (Cephalopoda). Histochemical localization and characterization of a specific high salt-soluble and heparin-soluble fraction of globular forms - Talesa_1995_Neurochem.Int_27_201 |
| Author(s) : Talesa V , Grauso M , Giovannini E , Rosi G , Toutant JP |
| Ref : Neurochem Int , 27 :201 , 1995 |
| Abstract : Talesa_1995_Neurochem.Int_27_201 |
| ESTHER : Talesa_1995_Neurochem.Int_27_201 |
| PubMedSearch : Talesa_1995_Neurochem.Int_27_201 |
| PubMedID: 7580876 |
| Title : Prophylactic transdermal treatment with physostigmine and scopolamine against soman intoxication in guinea-pigs - Meshulam_1995_J.Appl.Toxicol_15_263 |
| Author(s) : Meshulam Y , Davidovici R , Wengier A , Levy A |
| Ref : Journal of Applied Toxicology , 15 :263 , 1995 |
| Abstract : Meshulam_1995_J.Appl.Toxicol_15_263 |
| ESTHER : Meshulam_1995_J.Appl.Toxicol_15_263 |
| PubMedSearch : Meshulam_1995_J.Appl.Toxicol_15_263 |
| PubMedID: 7594194 |
| Title : Comparison of the effects of natural and synthetic huperzine-A on rat brain cholinergic function in vitro and in vivo - Tang_1994_J.Ethnopharmacol_44_147 |
| Author(s) : Tang XC , Kindel GH , Kozikowski AP , Hanin I |
| Ref : J Ethnopharmacol , 44 :147 , 1994 |
| Abstract : Tang_1994_J.Ethnopharmacol_44_147 |
| ESTHER : Tang_1994_J.Ethnopharmacol_44_147 |
| PubMedSearch : Tang_1994_J.Ethnopharmacol_44_147 |
| PubMedID: 7898122 |
| Title : [A kinetic study of the blood serum cholinesterase activity in the freshwater fish Abramis ballerus] - Zhukovskii_1994_Zh.Evol.Biokhim.Fiziol_30_177 |
| Author(s) : Zhukovskii Iu G , Kutsenko SA , Kuznetsova LP , Sochilina EE , Dmitrieva EN , Fartseiger NL , Tonkopii VD , Korkishko NN , Kozlovskaia VI , Fel'd VE |
| Ref : Zh Evol Biokhim Fiziol , 30 :177 , 1994 |
| Abstract : Zhukovskii_1994_Zh.Evol.Biokhim.Fiziol_30_177 |
| ESTHER : Zhukovskii_1994_Zh.Evol.Biokhim.Fiziol_30_177 |
| PubMedSearch : Zhukovskii_1994_Zh.Evol.Biokhim.Fiziol_30_177 |
| PubMedID: 7817653 |
| Title : [Aminostigmine as a cholinesterase inhibitor and as an agent for treating poisonings by cholinergic blockers]. [Russian] - Prozorovskii_1994_Eksp.Klin.Farmakol_57_13 |
| Author(s) : Prozorovskii VB , Livanov GA , Velikova VD , Afanas'ev VV , Pavlova LV |
| Ref : Eksperimentalnaia i Klinicheskaia Farmakologiia , 57 :13 , 1994 |
| Abstract : Prozorovskii_1994_Eksp.Klin.Farmakol_57_13 |
| ESTHER : Prozorovskii_1994_Eksp.Klin.Farmakol_57_13 |
| PubMedSearch : Prozorovskii_1994_Eksp.Klin.Farmakol_57_13 |
| PubMedID: 7696893 |
| Title : The molecular forms of acetylcholinesterase from Necator americanus (Nematoda), a hookworm parasite of the human intestine - Pritchard_1994_Eur.J.Biochem_219_317 |
| Author(s) : Pritchard DI , Brown A , Toutant JP |
| Ref : European Journal of Biochemistry , 219 :317 , 1994 |
| Abstract : Pritchard_1994_Eur.J.Biochem_219_317 |
| ESTHER : Pritchard_1994_Eur.J.Biochem_219_317 |
| PubMedSearch : Pritchard_1994_Eur.J.Biochem_219_317 |
| PubMedID: 8306998 |
| Title : Cholinesterase inhibitor effects on neurotransmitters in rat cortex in vivo - Cuadra_1994_J.Pharmacol.Exp.Ther_270_277 |
| Author(s) : Cuadra G , Summers K , Giacobini E |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 270 :277 , 1994 |
| Abstract : Cuadra_1994_J.Pharmacol.Exp.Ther_270_277 |
| ESTHER : Cuadra_1994_J.Pharmacol.Exp.Ther_270_277 |
| PubMedSearch : Cuadra_1994_J.Pharmacol.Exp.Ther_270_277 |
| PubMedID: 7913496 |
| Title : Acetylcholinesterase and butyrylcholinesterase expression in adult rabbit tissues and during development - Jbilo_1994_Eur.J.Biochem_225_115 |
| Author(s) : Jbilo O , L'Hermite Y , Talesa V , Toutant JP , Chatonnet A |
| Ref : European Journal of Biochemistry , 225 :115 , 1994 |
| Abstract : Jbilo_1994_Eur.J.Biochem_225_115 |
| ESTHER : Jbilo_1994_Eur.J.Biochem_225_115 |
| PubMedSearch : Jbilo_1994_Eur.J.Biochem_225_115 |
| PubMedID: 7925428 |
| Gene_locus related to this paper: rabit-ACHE , rabit-BCHE |
| Title : The effects of physostigmine and scopolamine on memory for food caches in the black-capped chickadee - Mineau_1994_Pharmacol.Biochem.Behav_49_363 |
| Author(s) : Mineau P , Boag PT , Beninger RJ |
| Ref : Pharmacology, Biochemistry & Behavior , 49 :363 , 1994 |
| Abstract : Mineau_1994_Pharmacol.Biochem.Behav_49_363 |
| ESTHER : Mineau_1994_Pharmacol.Biochem.Behav_49_363 |
| PubMedSearch : Mineau_1994_Pharmacol.Biochem.Behav_49_363 |
| PubMedID: 7824551 |
| Title : Tissue distribution of human acetylcholinesterase and butyrylcholinesterase messenger RNA - Jbilo_1994_Toxicon_32_1445 |
| Author(s) : Jbilo O , Bartels CF , Chatonnet A , Toutant JP , Lockridge O |
| Ref : Toxicon , 32 :1445 , 1994 |
| Abstract : Jbilo_1994_Toxicon_32_1445 |
| ESTHER : Jbilo_1994_Toxicon_32_1445 |
| PubMedSearch : Jbilo_1994_Toxicon_32_1445 |
| PubMedID: 7886701 |
| Title : Continuous administration of low dose rates of physostigmine and hyoscine to guinea-pigs prevents the toxicity and reduces the incapacitation produced by soman poisoning - Wetherell_1994_J.Pharm.Pharmacol_46_1023 |
| Author(s) : Wetherell JR |
| Ref : J Pharm Pharmacol , 46 :1023 , 1994 |
| Abstract : Wetherell_1994_J.Pharm.Pharmacol_46_1023 |
| ESTHER : Wetherell_1994_J.Pharm.Pharmacol_46_1023 |
| PubMedSearch : Wetherell_1994_J.Pharm.Pharmacol_46_1023 |
| PubMedID: 7714714 |
| Title : Solubilization and biochemical characterization of the melatonin deacetylase from Xenopus laevis retina - Grace_1993_J.Neurochem_60_990 |
| Author(s) : Grace MS , Besharse JC |
| Ref : Journal of Neurochemistry , 60 :990 , 1993 |
| Abstract : Grace_1993_J.Neurochem_60_990 |
| ESTHER : Grace_1993_J.Neurochem_60_990 |
| PubMedSearch : Grace_1993_J.Neurochem_60_990 |
| PubMedID: 8436983 |
| Title : Differentiation of esterases reacting with organophosphorus compounds - Reiner_1993_Chem.Biol.Interact_87_77 |
| Author(s) : Reiner E , Pavkovic E , Radic Z , Simeon-Rudolf V |
| Ref : Chemico-Biological Interactions , 87 :77 , 1993 |
| Abstract : Reiner_1993_Chem.Biol.Interact_87_77 |
| ESTHER : Reiner_1993_Chem.Biol.Interact_87_77 |
| PubMedSearch : Reiner_1993_Chem.Biol.Interact_87_77 |
| PubMedID: 8393750 |
| Title : Wuchereria bancrofti: identification of parasitic acetylcholinesterase in microfilariae infected human serum - Misra_1993_Trop.Med.Parasitol_44_75 |
| Author(s) : Misra S , Mohapatra TM , Rathaur S |
| Ref : Trop Med Parasitol , 44 :75 , 1993 |
| Abstract : Misra_1993_Trop.Med.Parasitol_44_75 |
| ESTHER : Misra_1993_Trop.Med.Parasitol_44_75 |
| PubMedSearch : Misra_1993_Trop.Med.Parasitol_44_75 |
| PubMedID: 8367669 |
| Title : Acetylcholinesterase activity is associated with efferent endings in the sensory epithelia of the utricle and semicircular canals of the rainbow trout inner ear - Khan_1993_Anat.Rec_237_141 |
| Author(s) : Khan KM , Drescher MJ , Hatfield JS , Drescher DG |
| Ref : Anatomical Record , 237 :141 , 1993 |
| Abstract : Khan_1993_Anat.Rec_237_141 |
| ESTHER : Khan_1993_Anat.Rec_237_141 |
| PubMedSearch : Khan_1993_Anat.Rec_237_141 |
| PubMedID: 8214639 |
| Title : Triiodothyronine (T3) modifies cholinergic-induced hypothermia and tremor in rats - Almeida_1993_Pharmacol.Biochem.Behav_46_729 |
| Author(s) : Almeida OM , Santos R |
| Ref : Pharmacology, Biochemistry & Behavior , 46 :729 , 1993 |
| Abstract : Almeida_1993_Pharmacol.Biochem.Behav_46_729 |
| ESTHER : Almeida_1993_Pharmacol.Biochem.Behav_46_729 |
| PubMedSearch : Almeida_1993_Pharmacol.Biochem.Behav_46_729 |
| PubMedID: 8278452 |
| Title : [The nervous system of Fibricola seoulensis by acetylcholinesterase histochemistry]. [Korean] - Cheon_1993_Korean.J.Parasitol_31_321 |
| Author(s) : Cheon EW , Kim CH |
| Ref : Korean Journal of Parasitology , 31 :321 , 1993 |
| Abstract : Cheon_1993_Korean.J.Parasitol_31_321 |
| ESTHER : Cheon_1993_Korean.J.Parasitol_31_321 |
| PubMedSearch : Cheon_1993_Korean.J.Parasitol_31_321 |
| PubMedID: 8297889 |
| Title : Esterases in quail (Coturnix coturnix). Physicochemical and developmental aspects - Tsitsiloni_1993_Comp.Biochem.Physiol.B_106_1009 |
| Author(s) : Tsitsiloni OE , Veini M , Haritos AA |
| Ref : Comparative Biochemistry & Physiology B Biochem Mol Biol , 106 :1009 , 1993 |
| Abstract : Tsitsiloni_1993_Comp.Biochem.Physiol.B_106_1009 |
| ESTHER : Tsitsiloni_1993_Comp.Biochem.Physiol.B_106_1009 |
| PubMedSearch : Tsitsiloni_1993_Comp.Biochem.Physiol.B_106_1009 |
| PubMedID: 8299343 |
| Title : Concurrent acute exercise alters central and peripheral responses to physostigmine - Dube_1993_Pharmacol.Biochem.Behav_46_827 |
| Author(s) : Dube SN , Somani SM , Babu SR |
| Ref : Pharmacology, Biochemistry & Behavior , 46 :827 , 1993 |
| Abstract : Dube_1993_Pharmacol.Biochem.Behav_46_827 |
| ESTHER : Dube_1993_Pharmacol.Biochem.Behav_46_827 |
| PubMedSearch : Dube_1993_Pharmacol.Biochem.Behav_46_827 |
| PubMedID: 8309962 |
| Title : Histochemical distribution of esterases in adult Onchocerca fasciata (Filarioidea: Onchocercidae) - Omar_1993_Trop.Med.Parasitol_4_295 |
| Author(s) : Omar MS , Raoof AM , Al-Amari OM |
| Ref : Trop Med Parasitol , 44 :295 , 1993 |
| Abstract : Omar_1993_Trop.Med.Parasitol_4_295 |
| ESTHER : Omar_1993_Trop.Med.Parasitol_4_295 |
| PubMedSearch : Omar_1993_Trop.Med.Parasitol_4_295 |
| PubMedID: 8134770 |
| Title : Cholinesterase inhibitor effects on extracellular acetylcholine in rat cortex - Messamore_1993_Neuropharmacol_32_745 |
| Author(s) : Messamore E , Warpman U , Ogane N , Giacobini E |
| Ref : Neuropharmacology , 32 :745 , 1993 |
| Abstract : Messamore_1993_Neuropharmacol_32_745 |
| ESTHER : Messamore_1993_Neuropharmacol_32_745 |
| PubMedSearch : Messamore_1993_Neuropharmacol_32_745 |
| PubMedID: 8413838 |
| Title : Phenserine: a physostigmine derivative that is a long-acting inhibitor of cholinesterase and demonstrates a wide dose range for attenuating a scopolamine-induced learning impairment of rats in a 14-unit T-maze - Iijima_1993_Psychopharmacology_112_415 |
| Author(s) : Iijima S , Greig NH , Garofalo P , Spangler EL , Heller B , Brossi A , Ingram DK |
| Ref : Psychopharmacology , 112 :415 , 1993 |
| Abstract : Iijima_1993_Psychopharmacology_112_415 |
| ESTHER : Iijima_1993_Psychopharmacology_112_415 |
| PubMedSearch : Iijima_1993_Psychopharmacology_112_415 |
| PubMedID: 7871051 |
| Title : Acetylcholinesterases of the nematode Steinernema carpocapsae. Characterization of two types of amphiphilic forms differing in their mode of membrane association - Arpagaus_1992_Eur.J.Biochem_207_1101 |
| Author(s) : Arpagaus M , Richier P , Berge JB , Toutant JP |
| Ref : European Journal of Biochemistry , 207 :1101 , 1992 |
| Abstract : Arpagaus_1992_Eur.J.Biochem_207_1101 |
| ESTHER : Arpagaus_1992_Eur.J.Biochem_207_1101 |
| PubMedSearch : Arpagaus_1992_Eur.J.Biochem_207_1101 |
| PubMedID: 1323459 |
| Title : In vitro protection of acetylcholinesterase and butyrylcholinesterase by tetrahydroaminoacridine. Comparison with physostigmine - Galli_1992_Biochem.Pharmacol_43_2427 |
| Author(s) : Galli A , Mori F , Gori I , Lucherini M |
| Ref : Biochemical Pharmacology , 43 :2427 , 1992 |
| Abstract : Galli_1992_Biochem.Pharmacol_43_2427 |
| ESTHER : Galli_1992_Biochem.Pharmacol_43_2427 |
| PubMedSearch : Galli_1992_Biochem.Pharmacol_43_2427 |
| PubMedID: 1610407 |
| Title : Preferential inhibition of acetylcholinesterase molecular forms in rat brain - Ogane_1992_Neurochem.Res_17_489 |
| Author(s) : Ogane N , Giacobini E , Messamore E |
| Ref : Neurochemical Research , 17 :489 , 1992 |
| Abstract : Ogane_1992_Neurochem.Res_17_489 |
| ESTHER : Ogane_1992_Neurochem.Res_17_489 |
| PubMedSearch : Ogane_1992_Neurochem.Res_17_489 |
| PubMedID: 1528356 |
| Title : Vascular effects of acetylcholinesterase inhibitors in the rabbit eye: a study with fasciculin and physostigmine - Silveira_1992_J.Ocul.Pharmacol_8_129 |
| Author(s) : Silveira R , Stjernschantz J |
| Ref : J Ocul Pharmacol , 8 :129 , 1992 |
| Abstract : Silveira_1992_J.Ocul.Pharmacol_8_129 |
| ESTHER : Silveira_1992_J.Ocul.Pharmacol_8_129 |
| PubMedSearch : Silveira_1992_J.Ocul.Pharmacol_8_129 |
| PubMedID: 1506754 |
| Title : Effects of combined acetylcholinesterase inhibition and serotonergic receptor blockade on age-associated memory impairments in rats - Normile_1992_Neurobiol.Aging_13_735 |
| Author(s) : Normile HJ , Altman HJ |
| Ref : Neurobiology of Aging , 13 :735 , 1992 |
| Abstract : Normile_1992_Neurobiol.Aging_13_735 |
| ESTHER : Normile_1992_Neurobiol.Aging_13_735 |
| PubMedSearch : Normile_1992_Neurobiol.Aging_13_735 |
| PubMedID: 1491739 |
| Title : Physostigmine (alone and together with adjunct) pretreatment against soman, sarin, tabun and VX intoxication - Harris_1991_Drug.Chem.Toxicol_14_265 |
| Author(s) : Harris LW , Talbot BG , Lennox WJ , Anderson DR , Solana RP |
| Ref : Drug & Chemical Toxicology , 14 :265 , 1991 |
| Abstract : Harris_1991_Drug.Chem.Toxicol_14_265 |
| ESTHER : Harris_1991_Drug.Chem.Toxicol_14_265 |
| PubMedSearch : Harris_1991_Drug.Chem.Toxicol_14_265 |
| PubMedID: 1935706 |
| Title : Effects of subacute pretreatment with carbamate together with acute adjunct pretreatment against nerve agent exposure - Anderson_1991_Drug.Chem.Toxicol_14_1 |
| Author(s) : Anderson DR , Harris LW , Lennox WJ , Solana RP |
| Ref : Drug & Chemical Toxicology , 14 :1 , 1991 |
| Abstract : Anderson_1991_Drug.Chem.Toxicol_14_1 |
| ESTHER : Anderson_1991_Drug.Chem.Toxicol_14_1 |
| PubMedSearch : Anderson_1991_Drug.Chem.Toxicol_14_1 |
| PubMedID: 1889370 |
| Title : Genetic variants of human serum cholinesterase influence metabolism of the muscle relaxant succinylcholine. - Lockridge_1990_Pharmacol.Ther_47_35 |
| Author(s) : Lockridge O |
| Ref : Pharmacol Ther , 47 :35 , 1990 |
| Abstract : Lockridge_1990_Pharmacol.Ther_47_35 |
| ESTHER : Lockridge_1990_Pharmacol.Ther_47_35 |
| PubMedSearch : Lockridge_1990_Pharmacol.Ther_47_35 |
| PubMedID: 2195556 |
| Gene_locus related to this paper: human-BCHE |
| Title : Pharmacokinetics and pharmacodynamics of physostigmine in the rat after oral administration - Somani_1989_Biopharm.Drug.Dispos_10_187 |
| Author(s) : Somani SM |
| Ref : Biopharmaceutics & Drug Disposition , 10 :187 , 1989 |
| Abstract : Somani_1989_Biopharm.Drug.Dispos_10_187 |
| ESTHER : Somani_1989_Biopharm.Drug.Dispos_10_187 |
| PubMedSearch : Somani_1989_Biopharm.Drug.Dispos_10_187 |
| PubMedID: 2706318 |
| Title : Comparative inhibitory effects of various physostigmine analogs against acetyl- and butyrylcholinesterases - Atack_1989_J.Pharmacol.Exp.Ther_249_194 |
| Author(s) : Atack JR , Yu QS , Soncrant TT , Brossi A , Rapoport SI |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 249 :194 , 1989 |
| Abstract : Atack_1989_J.Pharmacol.Exp.Ther_249_194 |
| ESTHER : Atack_1989_J.Pharmacol.Exp.Ther_249_194 |
| PubMedSearch : Atack_1989_J.Pharmacol.Exp.Ther_249_194 |
| PubMedID: 2709330 |
| Title : The relationship between oxime-induced reactivation of carbamylated acetylcholinesterase and antidotal efficacy against carbamate intoxication - Harris_1989_Toxicol.Appl.Pharmacol_98_128 |
| Author(s) : Harris LW , Talbot BG , Lennox WJ , Anderson DR |
| Ref : Toxicology & Applied Pharmacology , 98 :128 , 1989 |
| Abstract : Harris_1989_Toxicol.Appl.Pharmacol_98_128 |
| ESTHER : Harris_1989_Toxicol.Appl.Pharmacol_98_128 |
| PubMedSearch : Harris_1989_Toxicol.Appl.Pharmacol_98_128 |
| PubMedID: 2494778 |
| Title : An investigation of the in vitro inhibition of acetylcholinesterase by the carbamate inhibitor eserine sulphate in eserine resistant strains of Drosophila melanogaster - Burnell_1988_Comp.Biochem.Physiol.C_90_215 |
| Author(s) : Burnell AM , Wilkins NP |
| Ref : Comparative Biochemistry & Physiology C , 90 :215 , 1988 |
| Abstract : Burnell_1988_Comp.Biochem.Physiol.C_90_215 |
| ESTHER : Burnell_1988_Comp.Biochem.Physiol.C_90_215 |
| PubMedSearch : Burnell_1988_Comp.Biochem.Physiol.C_90_215 |
| PubMedID: 2904861 |
| Title : Interaction between bambuterol and physostigmine: aspects on cholinesterase inhibition and neuromuscular transmission in the smooth and skeletal muscles of the guinea-pig - Jeppsson_1988_Pharmacol.Toxicol_63_211 |
| Author(s) : Jeppsson AB , Waldeck B |
| Ref : Pharmacol Toxicol , 63 :211 , 1988 |
| Abstract : Jeppsson_1988_Pharmacol.Toxicol_63_211 |
| ESTHER : Jeppsson_1988_Pharmacol.Toxicol_63_211 |
| PubMedSearch : Jeppsson_1988_Pharmacol.Toxicol_63_211 |
| PubMedID: 2848230 |
| Title : Cholinesterases in rabbit serum - Simeon_1988_Gen.Pharmacol_19_849 |
| Author(s) : Simeon-Rudolf V , Reiner E , Skrinjaric-Spoljar M , Krauthacker B |
| Ref : General Pharmacology , 19 :849 , 1988 |
| Abstract : Simeon_1988_Gen.Pharmacol_19_849 |
| ESTHER : Simeon_1988_Gen.Pharmacol_19_849 |
| PubMedSearch : Simeon_1988_Gen.Pharmacol_19_849 |
| PubMedID: 3229626 |
| Title : A peptidase activity exhibited by human serum pseudocholinesterase - Boopathy_1987_Eur.J.Biochem_162_191 |
| Author(s) : Boopathy R , Balasubramanian AS |
| Ref : European Journal of Biochemistry , 162 :191 , 1987 |
| Abstract : Boopathy_1987_Eur.J.Biochem_162_191 |
| ESTHER : Boopathy_1987_Eur.J.Biochem_162_191 |
| PubMedSearch : Boopathy_1987_Eur.J.Biochem_162_191 |
| PubMedID: 3545820 |
| Title : Pharmacokinetics and pharmacodynamics of physostigmine in the rat after intravenous administration - Somani_1987_Drug.Metab.Dispos_15_627 |
| Author(s) : Somani SM , Khalique A |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 15 :627 , 1987 |
| Abstract : Somani_1987_Drug.Metab.Dispos_15_627 |
| ESTHER : Somani_1987_Drug.Metab.Dispos_15_627 |
| PubMedSearch : Somani_1987_Drug.Metab.Dispos_15_627 |
| PubMedID: 2891478 |
| Title : Kinetic investigations into the interactions of aprophen with cholinesterases and a carboxylesterase - Rush_1986_Biochem.Pharmacol_35_4167 |
| Author(s) : Rush RS , Doctor BP , Wolfe AD |
| Ref : Biochemical Pharmacology , 35 :4167 , 1986 |
| Abstract : Rush_1986_Biochem.Pharmacol_35_4167 |
| ESTHER : Rush_1986_Biochem.Pharmacol_35_4167 |
| PubMedSearch : Rush_1986_Biochem.Pharmacol_35_4167 |
| PubMedID: 3098245 |
| Title : Distribution and pharmacokinetics of physostigmine in rat after intramuscular administration - Somani_1986_Fundam.Appl.Toxicol_6_327 |
| Author(s) : Somani SM , Khalique A |
| Ref : Fundamental & Applied Toxicology , 6 :327 , 1986 |
| Abstract : Somani_1986_Fundam.Appl.Toxicol_6_327 |
| ESTHER : Somani_1986_Fundam.Appl.Toxicol_6_327 |
| PubMedSearch : Somani_1986_Fundam.Appl.Toxicol_6_327 |
| PubMedID: 3699321 |
| Title : Single-mosquito test to determine genotypes with an acetylcholinesterase insensitive to inhibition to propoxur insecticide - Raymond_1985_J.Am.Mosquito.Control.Assoc_1_425 |
| Author(s) : Raymond M , Fournier D , Berge JB , Cuany A , Bride JM , Pasteur N |
| Ref : Journal of the American Mosquito Control Association , 1 :425 , 1985 |
| Abstract : Raymond_1985_J.Am.Mosquito.Control.Assoc_1_425 |
| ESTHER : Raymond_1985_J.Am.Mosquito.Control.Assoc_1_425 |
| PubMedSearch : Raymond_1985_J.Am.Mosquito.Control.Assoc_1_425 |
| PubMedID: 3880259 |
| Title : In-vitro and in-vivo protection of acetylcholinesterase by eseroline against inactivation by diisopropyl fluorophosphate and carbamates - Galli_1985_J.Pharm.Pharmacol_37_42 |
| Author(s) : Galli A , Malmberg-Aiello P , Renzi G , Bartolini A |
| Ref : J Pharm Pharmacol , 37 :42 , 1985 |
| Abstract : Galli_1985_J.Pharm.Pharmacol_37_42 |
| ESTHER : Galli_1985_J.Pharm.Pharmacol_37_42 |
| PubMedSearch : Galli_1985_J.Pharm.Pharmacol_37_42 |
| PubMedID: 2858526 |
| Title : Polymorphism of pseudocholinesterase in Torpedo marmorata tissues: comparative study of the catalytic and molecular properties of this enzyme with acetylcholinesterase - Toutant_1985_J.Neurochem_44_580 |
| Author(s) : Toutant JP , Massoulie J , Bon S |
| Ref : Journal of Neurochemistry , 44 :580 , 1985 |
| Abstract : Toutant_1985_J.Neurochem_44_580 |
| ESTHER : Toutant_1985_J.Neurochem_44_580 |
| PubMedSearch : Toutant_1985_J.Neurochem_44_580 |
| PubMedID: 2578181 |
| Title : The reversible cholinesterase inhibitor physostigmine has channel-blocking and agonist effects on the acetylcholine receptor-ion channel complex - Shaw_1985_Mol.Pharmacol_28_527 |
| Author(s) : Shaw KP , Aracava Y , Akaike A , Daly JW , Rickett DL , Albuquerque EX |
| Ref : Molecular Pharmacology , 28 :527 , 1985 |
| Abstract : Shaw_1985_Mol.Pharmacol_28_527 |
| ESTHER : Shaw_1985_Mol.Pharmacol_28_527 |
| PubMedSearch : Shaw_1985_Mol.Pharmacol_28_527 |
| PubMedID: 2417099 |
| Title : [Multiple molecular forms of esterases from grass aphids inhibitory identification and stereospecificity] - Volkova_1983_Biokhimiia_48_1634 |
| Author(s) : Volkova RI , Titova EV |
| Ref : Biokhimiia , 48 :1634 , 1983 |
| Abstract : Volkova_1983_Biokhimiia_48_1634 |
| ESTHER : Volkova_1983_Biokhimiia_48_1634 |
| PubMedSearch : Volkova_1983_Biokhimiia_48_1634 |
| PubMedID: 6639987 |
| Title : Efficacy and toxicity of drug combinations in treatment of physostigmine toxicosis - Klemm_1983_Toxicology_27_41 |
| Author(s) : Klemm WR |
| Ref : Toxicology , 27 :41 , 1983 |
| Abstract : Klemm_1983_Toxicology_27_41 |
| ESTHER : Klemm_1983_Toxicology_27_41 |
| PubMedSearch : Klemm_1983_Toxicology_27_41 |
| PubMedID: 6149635 |
| Title : Activation and blockade of cardiac muscarinic receptors by endogenous acetylcholine and cholinesterase inhibitors - Brown_1982_J.Pharmacol.Exp.Ther_223_20 |
| Author(s) : Brown JH , Wetzel GT , Dunlap J |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 223 :20 , 1982 |
| Abstract : Brown_1982_J.Pharmacol.Exp.Ther_223_20 |
| ESTHER : Brown_1982_J.Pharmacol.Exp.Ther_223_20 |
| PubMedSearch : Brown_1982_J.Pharmacol.Exp.Ther_223_20 |
| PubMedID: 6288918 |
| Title : Serotonin-sensitive aryl acylamidase activity of platelet acetylcholinesterase - Majumdar_1982_Biochem.Pharmacol_31_2319 |
| Author(s) : Majumdar R , George ST , Balasubramanian AS |
| Ref : Biochemical Pharmacology , 31 :2319 , 1982 |
| Abstract : Majumdar_1982_Biochem.Pharmacol_31_2319 |
| ESTHER : Majumdar_1982_Biochem.Pharmacol_31_2319 |
| PubMedSearch : Majumdar_1982_Biochem.Pharmacol_31_2319 |
| PubMedID: 7126246 |
| Title : The aryl acylamidases and their relationship to cholinesterases in human serum, erythrocyte and liver - George_1981_Eur.J.Biochem_121_177 |
| Author(s) : George ST , Balasubramanian AS |
| Ref : European Journal of Biochemistry , 121 :177 , 1981 |
| Abstract : George_1981_Eur.J.Biochem_121_177 |
| ESTHER : George_1981_Eur.J.Biochem_121_177 |
| PubMedSearch : George_1981_Eur.J.Biochem_121_177 |
| PubMedID: 7035166 |
| Title : Differential inhibition of plasma cholinesterase variants using the dibutyrate analogue of pancuronium bromide - Whittaker_1981_Hum.Hered_31_242 |
| Author(s) : Whittaker M , Britten JJ |
| Ref : Hum Hered , 31 :242 , 1981 |
| Abstract : Whittaker_1981_Hum.Hered_31_242 |
| ESTHER : Whittaker_1981_Hum.Hered_31_242 |
| PubMedSearch : Whittaker_1981_Hum.Hered_31_242 |
| PubMedID: 7287015 |
| Title : The identity of the serotonin-sensitive aryl acylamidase with acetylcholinesterase from human erythrocytes, sheep basal ganglia and electric eel - George_1980_Eur.J.Biochem_111_511 |
| Author(s) : George ST , Balasubramanian AS |
| Ref : European Journal of Biochemistry , 111 :511 , 1980 |
| Abstract : George_1980_Eur.J.Biochem_111_511 |
| ESTHER : George_1980_Eur.J.Biochem_111_511 |
| PubMedSearch : George_1980_Eur.J.Biochem_111_511 |
| PubMedID: 7460914 |
| Title : Isolation and characterization of a butyrylesterase from human erythrocytes - Axenfors_1979_Biochim.Biophys.Acta_570_74 |
| Author(s) : Axenfors B , Andersson I , Augustinsson KB |
| Ref : Biochimica & Biophysica Acta , 570 :74 , 1979 |
| Abstract : Axenfors_1979_Biochim.Biophys.Acta_570_74 |
| ESTHER : Axenfors_1979_Biochim.Biophys.Acta_570_74 |
| PubMedSearch : Axenfors_1979_Biochim.Biophys.Acta_570_74 |
| PubMedID: 486506 |
| Title : The inhibition of frog tissue cholinesterases and the influence of eserine and prostigmine on the action of acetylcholine on the frog heart - Majcen_1975_Biochem.Pharmacol_24_1599 |
| Author(s) : Majcen Z , Brzin M |
| Ref : Biochemical Pharmacology , 24 :1599 , 1975 |
| Abstract : Majcen_1975_Biochem.Pharmacol_24_1599 |
| ESTHER : Majcen_1975_Biochem.Pharmacol_24_1599 |
| PubMedSearch : Majcen_1975_Biochem.Pharmacol_24_1599 |
| PubMedID: 1081394 |
| Title : The synthesis and anti-acetylcholinesterase activities of (plus)-physostigmine and (plus)-physovenine - |
| Author(s) : Dale FJ , Robinson B |
| Ref : J Pharm Pharmacol , 22 :889 , 1970 |
| PubMedID: 4395510 |
| Title : Treatment of myasthenia gravis with physostigmine - |
| Author(s) : Walker MB |
| Ref : Lancet , 1 :1200 , 1934 |
| PubMedID: |
| Title : Remarks on the physiological action and therapeutic uses of Physostigma venenosum or the ordeal bean of Calabar - |
| Author(s) : Hudson JQA |
| Ref : Southern Med. Rec , 3 :705 , 1873 |
| PubMedID: |
| Title : Uber die Wirkung einiger Gifte auf die Nerven der Glandula Submaxillaris - |
| Author(s) : Heidenhain R |
| Ref : Pflugers Arch Gesamte Physiol Menschen Tiere , 5 :309 , 1872 |
| PubMedID: |
| Title : An experimental research on the antagonism between the actions of physostigma and atropia - |
| Author(s) : Fraser TR |
| Ref : Trans Roy Soc Edinb , 26 :529 , 1870 |
| PubMedID: |
| Title : On the Physiological Action of the Calabar Bean (Physostigma venenosum Balfour) - |
| Author(s) : Fraser TR |
| Ref : J Anat Physiol , 9 :323 , 1867 |
| PubMedID: |
| Title : Beobachtung uber die Wirkung des Calabar-extracts gegen Atropin-Vergiftung - |
| Author(s) : Kleinwachter L |
| Ref : Berl Klin Wschr , 1 :367 , 1864 |
| PubMedID: |
| Title : On the characters, actions and therapeutic uses of the ordeal bean of Calabar (Physostigma venenosum Balfour) - |
| Author(s) : Fraser TR |
| Ref : Edinburgh Med J , 9 :36 , 1863 |
| PubMedID: |
| Title : On the calabar bean as a new agent in opthalmic medicine - |
| Author(s) : Robertson DA |
| Ref : Edinburgh Med J , 8 :815 , 1863 |
| PubMedID: |