SN-38
active metabolite of Irinotecan a chemotherapeutic pro-drug approved for the treatment of advanced colorectal cancer. Also product of hydrolysis of NPC product of hydrolysis of Irinotecan by 9sphn-E93
General
Type : Drug,Derivative of Irinotecan,Not A\/B H target,Pro-Drug
Chemical_Nomenclature : (19S)-10,19-diethyl-7,19-dihydroxy-17-oxa-3,13-diazapentacyclo[11.8.0.02,11.04,9.015,20]henicosa-1(21),2,4(9),5,7,10,15(20)-heptaene-14,18-dione
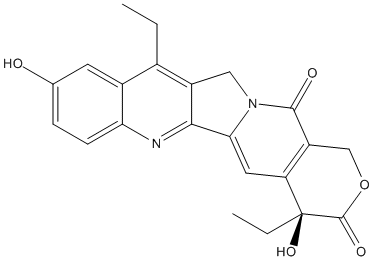
Canonical SMILES : CCC1=C2CN3C(=CC4=C(C3=O)COC(=O)C4(CC)O)C2=NC5=C1C=C(C=C5)O
InChI : InChI=1S\/C22H20N2O5\/c1-3-12-13-7-11(25)5-6-17(13)23-19-14(12)9-24-18(19)8-16-15(20(24)26)10-29-21(27)22(16,28)4-2\/h5-8,25,28H,3-4,9-10H2,1-2H3\/t22-\/m0\/s1
InChIKey : FJHBVJOVLFPMQE-QFIPXVFZSA-N
Other name(s) : 7-Ethyl-10-hydroxycamptothecin,CHEBI:8988,SCHEMBL34018,ZINC4099013,DB05482,RS4,SN-38,SN 38,SN38,SN 38 lactone
MW : 392.4
Formula : C22H20N2O5
CAS_number : 86639-52-3
PubChem : 104842
UniChem : FJHBVJOVLFPMQE-QFIPXVFZSA-N
IUPHAR :
Wikipedia :
Target
Families : SN-38 ligand of proteins in family: Carb_B_Bacteria || Carb_B_Chordata
Stucture :
Protein : 9sphn-E93 || human-CES2
References (27)
| Title : Structural insights into catalytical capability for CPT11 hydrolysis and substrate specificity of a novel marine microbial carboxylesterase, E93 - Li_2023_Front.Microbiol_13_1081094 |
| Author(s) : Li Y , Rong Z , Li Z , Cui H , Li J , Xu XW |
| Ref : Front Microbiol , 13 :1081094 , 2023 |
| Abstract : Li_2023_Front.Microbiol_13_1081094 |
| ESTHER : Li_2023_Front.Microbiol_13_1081094 |
| PubMedSearch : Li_2023_Front.Microbiol_13_1081094 |
| PubMedID: 36756200 |
| Gene_locus related to this paper: 9sphn-E93 |
| Title : Covalent CES2 Inhibitors Protect against Reduced Formation of Intestinal Organoids by the Anticancer Drug Irinotecan - Eades_2022_Curr.Drug.Metab__ |
| Author(s) : Eades W , Liu W , Shen Y , Shi Z , Yan B |
| Ref : Curr Drug Metab , : , 2022 |
| Abstract : Eades_2022_Curr.Drug.Metab__ |
| ESTHER : Eades_2022_Curr.Drug.Metab__ |
| PubMedSearch : Eades_2022_Curr.Drug.Metab__ |
| PubMedID: 36515038 |
| Title : Irinotecan-mediated diarrhea is mainly correlated with intestinal exposure to SN-38: Critical role of gut Ugt - Sun_2020_Toxicol.Appl.Pharmacol__115032 |
| Author(s) : Sun R , Zhu L , Li L , Song W , Gong X , Qi X , Wang Y , Ghose R , Gao S , Hu M , Liu Z |
| Ref : Toxicol Appl Pharmacol , :115032 , 2020 |
| Abstract : Sun_2020_Toxicol.Appl.Pharmacol__115032 |
| ESTHER : Sun_2020_Toxicol.Appl.Pharmacol__115032 |
| PubMedSearch : Sun_2020_Toxicol.Appl.Pharmacol__115032 |
| PubMedID: 32387182 |
| Title : Combined SN-38 and gefitinib treatment promotes CD44 degradation in head and neck squamous cell carcinoma cells - Nanbu_2018_Oncol.Rep_39_367 |
| Author(s) : Nanbu T , Umemura N , Ohkoshi E , Nanbu K , Sakagami H , Shimada J |
| Ref : Oncol Rep , 39 :367 , 2018 |
| Abstract : Nanbu_2018_Oncol.Rep_39_367 |
| ESTHER : Nanbu_2018_Oncol.Rep_39_367 |
| PubMedSearch : Nanbu_2018_Oncol.Rep_39_367 |
| PubMedID: 29192320 |
| Title : Acceleration of carboxylesterase-mediated activation of irinotecan to SN-38 by serum from patients with end-stage kidney disease - Koide_2018_Cancer.Chemother.Pharmacol_81_1121 |
| Author(s) : Koide H , Tsujimoto M , Katsube Y , Ochiai M , Hojo A , Furukubo T , Izumi S , Yamakawa T , Shima D , Minegaki T , Nishiguchi K |
| Ref : Cancer Chemother Pharmacol , 81 :1121 , 2018 |
| Abstract : Koide_2018_Cancer.Chemother.Pharmacol_81_1121 |
| ESTHER : Koide_2018_Cancer.Chemother.Pharmacol_81_1121 |
| PubMedSearch : Koide_2018_Cancer.Chemother.Pharmacol_81_1121 |
| PubMedID: 29693202 |
| Title : Pharmacokinetics and safety of DTS-108, a human oligopeptide bound to SN-38 with an esterase-sensitive cross-linker in patients with advanced malignancies: a Phase I study - Coriat_2016_Int.J.Nanomedicine_11_6207 |
| Author(s) : Coriat R , Faivre SJ , Mir O , Dreyer C , Ropert S , Bouattour M , Desjardins R , Goldwasser F , Raymond E |
| Ref : Int J Nanomedicine , 11 :6207 , 2016 |
| Abstract : Coriat_2016_Int.J.Nanomedicine_11_6207 |
| ESTHER : Coriat_2016_Int.J.Nanomedicine_11_6207 |
| PubMedSearch : Coriat_2016_Int.J.Nanomedicine_11_6207 |
| PubMedID: 27920527 |
| Title : Impact of obesity on accumulation of the toxic irinotecan metabolite, SN-38, in mice - Mallick_2015_Life.Sci_139_132 |
| Author(s) : Mallick P , Shah P , Gandhi A , Ghose R |
| Ref : Life Sciences , 139 :132 , 2015 |
| Abstract : Mallick_2015_Life.Sci_139_132 |
| ESTHER : Mallick_2015_Life.Sci_139_132 |
| PubMedSearch : Mallick_2015_Life.Sci_139_132 |
| PubMedID: 26334566 |
| Title : OATP1A\/1B Transporters Affect Irinotecan and SN-38 Pharmacokinetics and Carboxylesterase Expression in Knockout and Humanized Transgenic Mice - Iusuf_2014_Mol.Cancer.Ther_13_492 |
| Author(s) : Iusuf D , Ludwig M , Elbatsh A , van Esch A , van de Steeg E , Wagenaar E , van der Valk M , Lin F , van Tellingen O , Schinkel AH |
| Ref : Mol Cancer Ther , 13 :492 , 2014 |
| Abstract : Iusuf_2014_Mol.Cancer.Ther_13_492 |
| ESTHER : Iusuf_2014_Mol.Cancer.Ther_13_492 |
| PubMedSearch : Iusuf_2014_Mol.Cancer.Ther_13_492 |
| PubMedID: 24194565 |
| Title : Synthesis and Biological Evaluation of Novel 10-Substituted-7-ethyl-10-hydroxycamptothecin (SN-38) Prodrugs - Zhou_2014_Molecules_19_19718 |
| Author(s) : Zhou M , Liu M , He X , Yu H , Wu D , Yao Y , Fan S , Zhang P , Shi W , Zhong B |
| Ref : Molecules , 19 :19718 , 2014 |
| Abstract : Zhou_2014_Molecules_19_19718 |
| ESTHER : Zhou_2014_Molecules_19_19718 |
| PubMedSearch : Zhou_2014_Molecules_19_19718 |
| PubMedID: 25438082 |
| Title : Targeting colorectal cancer cells with single-walled carbon nanotubes conjugated to anticancer agent SN-38 and EGFR antibody - Lee_2013_Biomaterials_34_8756 |
| Author(s) : Lee PC , Chiou YC , Wong JM , Peng CL , Shieh MJ |
| Ref : Biomaterials , 34 :8756 , 2013 |
| Abstract : Lee_2013_Biomaterials_34_8756 |
| ESTHER : Lee_2013_Biomaterials_34_8756 |
| PubMedSearch : Lee_2013_Biomaterials_34_8756 |
| PubMedID: 23937913 |
| Title : A new UPLC-MS\/MS method for the determination of irinotecan and 7-ethyl-10-hydroxycamptothecin (SN-38) in mice: application to plasma and brain pharmacokinetics - Goldwirt_2012_J.Pharm.Biomed.Anal_66_325 |
| Author(s) : Goldwirt L , Lemaitre F , Zahr N , Farinotti R , Fernandez C |
| Ref : J Pharm Biomed Anal , 66 :325 , 2012 |
| Abstract : Goldwirt_2012_J.Pharm.Biomed.Anal_66_325 |
| ESTHER : Goldwirt_2012_J.Pharm.Biomed.Anal_66_325 |
| PubMedSearch : Goldwirt_2012_J.Pharm.Biomed.Anal_66_325 |
| PubMedID: 22551773 |
| Title : Magnetic-Fe\/Fe(3)O(4)-nanoparticle-bound SN38 as carboxylesterase-cleavable prodrug for the delivery to tumors within monocytes\/macrophages - Wang_2012_Beilstein.J.Nanotechnol_3_444 |
| Author(s) : Wang H , Shrestha TB , Basel MT , Dani RK , Seo GM , Balivada S , Pyle MM , Prock H , Koper OB , Thapa PS , Moore D , Li P , Chikan V , Troyer DL , Bossmann SH |
| Ref : Beilstein J Nanotechnol , 3 :444 , 2012 |
| Abstract : Wang_2012_Beilstein.J.Nanotechnol_3_444 |
| ESTHER : Wang_2012_Beilstein.J.Nanotechnol_3_444 |
| PubMedSearch : Wang_2012_Beilstein.J.Nanotechnol_3_444 |
| PubMedID: 23016149 |
| Title : Involvement of up-regulation of hepatic breast cancer resistance protein in decreased plasma concentration of 7-ethyl-10-hydroxycamptothecin (SN-38) by coadministration of S-1 in rats - Yokoo_2007_Drug.Metab.Dispos_35_1511 |
| Author(s) : Yokoo K , Hamada A , Watanabe H , Matsuzaki T , Imai T , Fujimoto H , Masa K , Saito H |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 35 :1511 , 2007 |
| Abstract : Yokoo_2007_Drug.Metab.Dispos_35_1511 |
| ESTHER : Yokoo_2007_Drug.Metab.Dispos_35_1511 |
| PubMedSearch : Yokoo_2007_Drug.Metab.Dispos_35_1511 |
| PubMedID: 17537871 |
| Title : Liposomalization of SN-38 as active metabolite of CPT-11 - Sadzuka_2005_J.Control.Release_108_453 |
| Author(s) : Sadzuka Y , Takabe H , Sonobe T |
| Ref : J Control Release , 108 :453 , 2005 |
| Abstract : Sadzuka_2005_J.Control.Release_108_453 |
| ESTHER : Sadzuka_2005_J.Control.Release_108_453 |
| PubMedSearch : Sadzuka_2005_J.Control.Release_108_453 |
| PubMedID: 16182400 |
| Title : Carboxylesterase isoform 2 mRNA expression in peripheral blood mononuclear cells is a predictive marker of the irinotecan to SN38 activation step in colorectal cancer patients - Cecchin_2005_Clin.Cancer.Res_11_6901 |
| Author(s) : Cecchin E , Corona G , Masier S , Biason P , Cattarossi G , Frustaci S , Buonadonna A , Colussi A , Toffoli G |
| Ref : Clin Cancer Research , 11 :6901 , 2005 |
| Abstract : Cecchin_2005_Clin.Cancer.Res_11_6901 |
| ESTHER : Cecchin_2005_Clin.Cancer.Res_11_6901 |
| PubMedSearch : Cecchin_2005_Clin.Cancer.Res_11_6901 |
| PubMedID: 16203781 |
| Title : In vitro conversion of irinotecan to SN-38 in human plasma - Shingyoji_2004_Cancer.Sci_95_537 |
| Author(s) : Shingyoji M , Takiguchi Y , Watanabe-Uruma R , Asaka-Amano Y , Matsubara H , Kurosu K , Kasahara Y , Tanabe N , Tatsumi K , Kuriyama T |
| Ref : Cancer Sci , 95 :537 , 2004 |
| Abstract : Shingyoji_2004_Cancer.Sci_95_537 |
| ESTHER : Shingyoji_2004_Cancer.Sci_95_537 |
| PubMedSearch : Shingyoji_2004_Cancer.Sci_95_537 |
| PubMedID: 15182436 |
| Title : Pharmacogenetics of human carboxylesterase 2, an enzyme involved in the activation of irinotecan into SN-38 - Charasson_2004_Clin.Pharmacol.Ther_76_528 |
| Author(s) : Charasson V , Bellott R , Meynard D , Longy M , Gorry P , Robert J |
| Ref : Clinical Pharmacology & Therapeutics , 76 :528 , 2004 |
| Abstract : Charasson_2004_Clin.Pharmacol.Ther_76_528 |
| ESTHER : Charasson_2004_Clin.Pharmacol.Ther_76_528 |
| PubMedSearch : Charasson_2004_Clin.Pharmacol.Ther_76_528 |
| PubMedID: 15592324 |
| Gene_locus related to this paper: human-CES2 |
| Title : A prodrug strategy using ONYX-015-based replicating adenoviruses to deliver rabbit carboxylesterase to tumor cells for conversion of CPT-11 to SN-38 - Stubdal_2003_Cancer.Res_63_6900 |
| Author(s) : Stubdal H , Perin N , Lemmon M , Holman P , Bauzon M , Potter PM , Danks MK , Fattaey A , Dubensky T , Johnson L |
| Ref : Cancer Research , 63 :6900 , 2003 |
| Abstract : Stubdal_2003_Cancer.Res_63_6900 |
| ESTHER : Stubdal_2003_Cancer.Res_63_6900 |
| PubMedSearch : Stubdal_2003_Cancer.Res_63_6900 |
| PubMedID: 14583489 |
| Title : Intracellular conversion of irinotecan to its active form, SN-38, by native carboxylesterase in human non-small cell lung cancer - Ohtsuka_2003_Lung.Cancer_41_187 |
| Author(s) : Ohtsuka K , Inoue S , Kameyama M , Kanetoshi A , Fujimoto T , Takaoka K , Araya Y , Shida A |
| Ref : Lung Cancer , 41 :187 , 2003 |
| Abstract : Ohtsuka_2003_Lung.Cancer_41_187 |
| ESTHER : Ohtsuka_2003_Lung.Cancer_41_187 |
| PubMedSearch : Ohtsuka_2003_Lung.Cancer_41_187 |
| PubMedID: 12871782 |
| Title : The relative contributions of carboxylesterase and beta-glucuronidase in the formation of SN-38 in human colorectal tumours - Tobin_2003_Oncol.Rep_10_1977 |
| Author(s) : Tobin PJ , Dodds HM , Clarke S , Schnitzler M , Rivory LP |
| Ref : Oncol Rep , 10 :1977 , 2003 |
| Abstract : Tobin_2003_Oncol.Rep_10_1977 |
| ESTHER : Tobin_2003_Oncol.Rep_10_1977 |
| PubMedSearch : Tobin_2003_Oncol.Rep_10_1977 |
| PubMedID: 14534729 |
| Title : Human plasma carboxylesterase and butyrylcholinesterase enzyme activity: correlations with SN-38 pharmacokinetics during a prolonged infusion of irinotecan - Guemei_2001_Cancer.Chemother.Pharmacol_47_283 |
| Author(s) : Guemei AA , Cottrell J , Band R , Hehman H , Prudhomme M , Pavlov MV , Grem JL , Ismail AS , Bowen D , Taylor RE , Takimoto CH |
| Ref : Cancer Chemother Pharmacol , 47 :283 , 2001 |
| Abstract : Guemei_2001_Cancer.Chemother.Pharmacol_47_283 |
| ESTHER : Guemei_2001_Cancer.Chemother.Pharmacol_47_283 |
| PubMedSearch : Guemei_2001_Cancer.Chemother.Pharmacol_47_283 |
| PubMedID: 11345644 |
| Title : The anticancer prodrug CPT-11 is a potent inhibitor of acetylcholinesterase but is rapidly catalyzed to SN-38 by butyrylcholinesterase - Morton_1999_Cancer.Res_59_1458 |
| Author(s) : Morton CL , Wadkins RM , Danks MK , Potter PM |
| Ref : Cancer Research , 59 :1458 , 1999 |
| Abstract : Morton_1999_Cancer.Res_59_1458 |
| ESTHER : Morton_1999_Cancer.Res_59_1458 |
| PubMedSearch : Morton_1999_Cancer.Res_59_1458 |
| PubMedID: 10197614 |
| Title : Conversion of the CPT-11 metabolite APC to SN-38 by rabbit liver carboxylesterase - Guichard_1998_Clin.Cancer.Res_4_3089 |
| Author(s) : Guichard SM , Morton CL , Krull EJ , Stewart CF , Danks MK , Potter PM |
| Ref : Clin Cancer Research , 4 :3089 , 1998 |
| Abstract : Guichard_1998_Clin.Cancer.Res_4_3089 |
| ESTHER : Guichard_1998_Clin.Cancer.Res_4_3089 |
| PubMedSearch : Guichard_1998_Clin.Cancer.Res_4_3089 |
| PubMedID: 9865925 |
| Title : Identification of a new metabolite of CPT-11 (irinotecan): pharmacological properties and activation to SN-38 - Dodds_1998_J.Pharmacol.Exp.Ther_286_578 |
| Author(s) : Dodds HM , Haaz MC , Riou JF , Robert J , Rivory LP |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 286 :578 , 1998 |
| Abstract : Dodds_1998_J.Pharmacol.Exp.Ther_286_578 |
| ESTHER : Dodds_1998_J.Pharmacol.Exp.Ther_286_578 |
| PubMedSearch : Dodds_1998_J.Pharmacol.Exp.Ther_286_578 |
| PubMedID: 9655905 |
| Title : The transformation of irinotecan (CPT-11) to its active metabolite SN-38 by human liver microsomes. Differential hydrolysis for the lactone and carboxylate forms - Haaz_1997_Naunyn.Schmiedebergs.Arch.Pharmacol_356_257 |
| Author(s) : Haaz MC , Rivory LP , Riche C , Robert J |
| Ref : Naunyn Schmiedebergs Arch Pharmacol , 356 :257 , 1997 |
| Abstract : Haaz_1997_Naunyn.Schmiedebergs.Arch.Pharmacol_356_257 |
| ESTHER : Haaz_1997_Naunyn.Schmiedebergs.Arch.Pharmacol_356_257 |
| PubMedSearch : Haaz_1997_Naunyn.Schmiedebergs.Arch.Pharmacol_356_257 |
| PubMedID: 9272733 |
| Title : Bioactivation of the anticancer agent CPT-11 to SN-38 by human hepatic microsomal carboxylesterases and the in vitro assessment of potential drug interactions - Slatter_1997_Drug.Metab.Dispos_25_1157 |
| Author(s) : Slatter JG , Su P , Sams JP , Schaaf LJ , Wienkers LC |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 25 :1157 , 1997 |
| Abstract : Slatter_1997_Drug.Metab.Dispos_25_1157 |
| ESTHER : Slatter_1997_Drug.Metab.Dispos_25_1157 |
| PubMedSearch : Slatter_1997_Drug.Metab.Dispos_25_1157 |
| PubMedID: 9321519 |
| Title : Conversion of irinotecan (CPT-11) to its active metabolite, 7-ethyl-10-hydroxycamptothecin (SN-38), by human liver carboxylesterase - Rivory_1996_Biochem.Pharmacol_52_1103 |
| Author(s) : Rivory LP , Bowles MR , Robert J , Pond SM |
| Ref : Biochemical Pharmacology , 52 :1103 , 1996 |
| Abstract : Rivory_1996_Biochem.Pharmacol_52_1103 |
| ESTHER : Rivory_1996_Biochem.Pharmacol_52_1103 |
| PubMedSearch : Rivory_1996_Biochem.Pharmacol_52_1103 |
| PubMedID: 8831730 |