HI-6
General
Type : Oxime, Bispyridinium, Carboxamide, Hagedorn Oxime
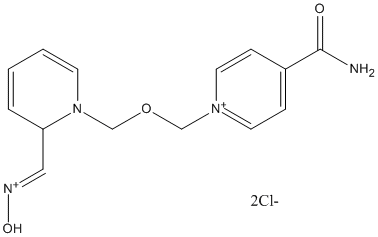
Chemical_Nomenclature : [(E)-[1-[(4-carbamoylpyridin-1-ium-1-yl)methoxymethyl]pyridin-2-ylidene]methyl]-oxoazanium\;dichloride, 1-[[2-[(E)-hydroxyiminomethyl]pyridin-1-ium-1-yl]methoxymethyl]pyridin-1-ium-4-carboxamide\;dichloride, 1-[[2-[(Z)-hydroxyiminomethyl]pyridin-1-ium-1-yl]methoxymethyl]pyridin-1-ium-4-carboxamide\;methanesulfonate
Canonical SMILES : C1=CC(=C[NH+]=O)N(C=C1)COC[N+]2=CC=C(C=C2)C(=O)N.[Cl-].[Cl-] || C1=CC=[N+](C(=C1)C=NO)COC[N+]2=CC=C(C=C2)C(=O)N.[Cl-].[Cl-] || CS(=O)(=O)[O-].CS(=O)(=O)[O-].C1=CC=[N+](C(=C1)C=NO)COC[N+]2=CC=C(C=C2)C(=O)N
InChI : InChI=1S\/C14H14N4O3.2ClH\/c15-14(19)12-4-7-17(8-5-12)10-21-11-18-6-2-1-3-13(18)9-16-20\;\;\/h1-9H,10-11H2,(H-,15,19)\;2*1H\/b13-9+\;\;, InChI=1S\/C14H14N4O3.2ClH\/c15-14(19)12-4-7-17(8-5-12)10-21-11-18-6-2-1-3-13(18)9-16-20\;\;\/h1-9H,10-11H2,(H-,15,19)\;2*1H, InChI=1S\/C14H14N4O3.2CH4O3S\/c15-14(19)12-4-7-17(8-5-12)10-21-11-18-6-2-1-3-13(18)9-16-20\;2*1-5(2,3)4\/h1-9H,10-11H2,(H-,15,19)\;2*1H3,(H,2,3,4)
InChIKey : QELSIJXWEROXOE-IGUOPLJTSA-N, QELSIJXWEROXOE-UHFFFAOYSA-N, KSZUKDJWBYNHBO-UHFFFAOYSA-N
Other name(s) : 1-(((4-(aminocarbonyl)pyridinio)methoxy)methyl)-2-((hydr oxyimino)methyl pyridinium dichloride monohydrate 1,1'-[oxybis(methylene)]-[4-carbamoyl-2'-(hydroxyimino)methyl]pyridinium dichloride, Asoxime chloride, Asoxime, CCRIS 7699, HJ 6, HI 6, HI6, UNII-HUV88P6SJS, HUV88P6SJS, HI-6 Dichloride, Asoxime (dichloride), SCHEMBL667540, SCHEMBL840733, SCHEMBL20917382, Asoxime dimethanesulfonate, Q27290555
MW : 359.20 || 478.5
Formula : C14H16Cl2N4O3 || C16H22N4O9S2
CAS_number : 34433-31-3
CID PubChem :
InChIKey UniChem :
Iuphar :
Wikipedia : Asoxime_chloride
References
No reference