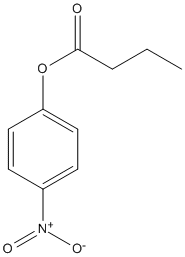
Paranitrophenylbutyrate
General
Type : pNP || Chromogen || Butyrate || Water-soluble short chain ester
Chemical_Nomenclature : (4-nitrophenyl) butanoate
Canonical SMILES : CCCC(=O)OC1=CC=C(C=C1)[N+](=O)[O-]
InChI : InChI=1S\/C10H11NO4\/c1-2-3-10(12)15-9-6-4-8(5-7-9)11(13)14\/h4-7H,2-3H2,1H3
InChIKey : DVDUMIQZEUTAGK-UHFFFAOYSA-N
Other name(s) : pNP-C4, pNP-butyrate, pNP butyrate, pNPB, p-NPB, pNP4, p-NP butyrate, 4-Nitrophenyl butyrate, P-Nitrophenyl butyrate, P-Nitrophenol butyrate, Para-Nitrophenyl butyrate, P-nitrophenyl butanoate
Target
Families : Carb_B_Bacteria, Bacterial_lip_FamI.5, Canar_LipB, Lipase_3, GTSAGmotif, Duf_3089, HNLyase_Bact, BioH, Bacterial_Est97, Bacterial_EstLip_FamX, abh_upf00227, 6_AlphaBeta_hydrolase, LYsophospholipase_carboxylesterase, A85-EsteraseD-FGH, Duf_1100-S, Est9X, Polyesterase-lipase-cutinase, Fungal_carboxylesterase_lipase, Hormone-sensitive_lipase_like, Carb_B_Chordata, Cholesterol_esterase, AlphaBeta_hydrolase, ABHD11-Acetyl_transferase, Pectinacetylesterase-Notum, Cutinase, CarbLipBact_1, Bacterial_esterase, Bacterial_lip_FamI.1, Monoglyceridelipase_lysophospholip
References (19)
| Title : Cutinase-Catalyzed Polyester-Polyurethane Degradation: Elucidation of the Hydrolysis Mechanism - Di Bisceglie_2022_Polymers.(Basel)_14_411 |
| Author(s) : Di Bisceglie F , Quartinello F , Vielnascher R , Guebitz GM , Pellis A |
| Ref : Polymers (Basel) , 14 :411 , 2022 |
| Abstract : Di Bisceglie_2022_Polymers.(Basel)_14_411 |
| ESTHER : Di Bisceglie_2022_Polymers.(Basel)_14_411 |
| PubMedSearch : Di Bisceglie_2022_Polymers.(Basel)_14_411 |
| PubMedID: 35160402 |
| Gene_locus related to this paper: humin-cut |
| Title : Biochemical and Structural Characterization of a novel thermophilic esterase EstD11 provide catalytic insights for the HSL family - Miguel-Ruano_2021_Comput.Struct.Biotechnol.J_19_1214 |
| Author(s) : Miguel-Ruano V , Rivera I , Rajkovic J , Knapik K , Torrado A , Otero JM , Beneventi E , Becerra M , Sanchez-Costa M , Hidalgo A , Berenguer J , Gonzalez-Siso MI , Cruces J , Rua ML , Hermoso JA |
| Ref : Comput Struct Biotechnol J , 19 :1214 , 2021 |
| Abstract : Miguel-Ruano_2021_Comput.Struct.Biotechnol.J_19_1214 |
| ESTHER : Miguel-Ruano_2021_Comput.Struct.Biotechnol.J_19_1214 |
| PubMedSearch : Miguel-Ruano_2021_Comput.Struct.Biotechnol.J_19_1214 |
| PubMedID: 33680362 |
| Gene_locus related to this paper: 9delt-EstD11 |
| Title : Phylogenetic analysis and in-depth characterization of functionally and structurally diverse CE5 cutinases - Novy_2021_J.Biol.Chem__101302 |
| Author(s) : Novy V , Carneiro LV , Shin JH , Larsbrink J , Olsson L |
| Ref : Journal of Biological Chemistry , :101302 , 2021 |
| Abstract : Novy_2021_J.Biol.Chem__101302 |
| ESTHER : Novy_2021_J.Biol.Chem__101302 |
| PubMedSearch : Novy_2021_J.Biol.Chem__101302 |
| PubMedID: 34653507 |
| Gene_locus related to this paper: 9pezi-s4vch4 , aspfu-q4x1n0 , crysp-Q874E9 , emeni-q5b2c1 , fusso-cutas , pyrbr-Q9Y7G8 , copci-b9u443 , thite-g2rae6 , strsw-c9zcr8 |
| Title : Structure-guided engineering of a Thermobifida fusca cutinase for enhanced hydrolysis on natural polyester substrate - Dong_2020_Bioresour.Bioprocess_7_37 |
| Author(s) : Dong Q , Yuan S , Wu L , Su L , Zhao Q , Wu J , Huang W , Zhou J |
| Ref : Bioresour. Bioprocess , 7 :37 , 2020 |
| Abstract : Dong_2020_Bioresour.Bioprocess_7_37 |
| ESTHER : Dong_2020_Bioresour.Bioprocess_7_37 |
| PubMedSearch : Dong_2020_Bioresour.Bioprocess_7_37 |
| PubMedID: |
| Gene_locus related to this paper: thefu-q6a0i4 |
| Title : A novel live cell assay to measure diacylglycerol lipase alpha activity - Singh_2016_Biosci.Rep_36_ |
| Author(s) : Singh PK , Markwick R , Howell FV , Williams G , Doherty P |
| Ref : Bioscience Reports , 36 : , 2016 |
| Abstract : Singh_2016_Biosci.Rep_36_ |
| ESTHER : Singh_2016_Biosci.Rep_36_ |
| PubMedSearch : Singh_2016_Biosci.Rep_36_ |
| PubMedID: 27013337 |
| Gene_locus related to this paper: human-DAGLA |
| Title : Characterization of LipN (Rv2970c) of Mycobacterium Tuberculosis H37Rv and its Probable Role in Xenobiotic Degradation - Jadeja_2016_J.Cell.Biochem_117_390 |
| Author(s) : Jadeja D , Dogra N , Arya S , Singh G , Kaur J |
| Ref : Journal of Cellular Biochemistry , 117 :390 , 2016 |
| Abstract : Jadeja_2016_J.Cell.Biochem_117_390 |
| ESTHER : Jadeja_2016_J.Cell.Biochem_117_390 |
| PubMedSearch : Jadeja_2016_J.Cell.Biochem_117_390 |
| PubMedID: 26212120 |
| Gene_locus related to this paper: myctu-Rv2970c |
| Title : Structural insights of a hormone sensitive lipase homologue Est22 - Huang_2016_Sci.Rep_6_28550 |
| Author(s) : Huang J , Huo YY , Ji R , Kuang S , Ji C , Xu XW , Li J |
| Ref : Sci Rep , 6 :28550 , 2016 |
| Abstract : Huang_2016_Sci.Rep_6_28550 |
| ESTHER : Huang_2016_Sci.Rep_6_28550 |
| PubMedSearch : Huang_2016_Sci.Rep_6_28550 |
| PubMedID: 27328716 |
| Gene_locus related to this paper: 9bact-H6BDX1 |
| Title : The Structure of a Novel Thermophilic Esterase from the Planctomycetes Species, Reveals an Open Active Site Due to a Minimal 'Cap' Domain - Sayer_2015_Front.Microbiol_6_1294 |
| Author(s) : Sayer C , Szabo Z , Isupov MN , Ingham C , Littlechild JA |
| Ref : Front Microbiol , 6 :1294 , 2015 |
| Abstract : Sayer_2015_Front.Microbiol_6_1294 |
| ESTHER : Sayer_2015_Front.Microbiol_6_1294 |
| PubMedSearch : Sayer_2015_Front.Microbiol_6_1294 |
| PubMedID: 26635762 |
| Gene_locus related to this paper: 9bact-TtEst2 |
| Title : Directed evolution of new and improved enzyme functions using an evolutionary intermediate and multidirectional search - Porter_2015_ACS.Chem.Biol_10_611 |
| Author(s) : Porter JL , Boon PL , Murray TP , Huber T , Collyer CA , Ollis DL |
| Ref : ACS Chemical Biology , 10 :611 , 2015 |
| Abstract : Porter_2015_ACS.Chem.Biol_10_611 |
| ESTHER : Porter_2015_ACS.Chem.Biol_10_611 |
| PubMedSearch : Porter_2015_ACS.Chem.Biol_10_611 |
| PubMedID: 25419863 |
| Gene_locus related to this paper: psepu-clcd1 |
| Title : Roles of tryptophan residue and disulfide bond in the variable lid region of oxidized polyvinyl alcohol hydrolase - Yang_2014_Biochem.Biophys.Res.Commun_452_509 |
| Author(s) : Yang Y , Ko TP , Liu L , Li J , Huang CH , Chen J , Guo RT , Du G |
| Ref : Biochemical & Biophysical Research Communications , 452 :509 , 2014 |
| Abstract : Yang_2014_Biochem.Biophys.Res.Commun_452_509 |
| ESTHER : Yang_2014_Biochem.Biophys.Res.Commun_452_509 |
| PubMedSearch : Yang_2014_Biochem.Biophys.Res.Commun_452_509 |
| PubMedID: 25173935 |
| Gene_locus related to this paper: sphs1-OPH |
| Title : SulE, a sulfonylurea herbicide de-esterification esterase from Hansschlegelia zhihuaiae S113 - Hang_2012_Appl.Environ.Microbiol_78_1962 |
| Author(s) : Hang BJ , Hong Q , Xie XT , Huang X , Wang CH , He J , Li SP |
| Ref : Applied Environmental Microbiology , 78 :1962 , 2012 |
| Abstract : Hang_2012_Appl.Environ.Microbiol_78_1962 |
| ESTHER : Hang_2012_Appl.Environ.Microbiol_78_1962 |
| PubMedSearch : Hang_2012_Appl.Environ.Microbiol_78_1962 |
| PubMedID: 22247165 |
| Gene_locus related to this paper: 9rhiz-g9i933 |
| Title : Non-lipolytic and lipolytic sequence-related carboxylesterases: a comparative study of the structure-function relationships of rabbit liver esterase 1 and bovine pancreatic bile-salt-activated lipase - Chahinian_2010_Biochim.Biophys.Acta_1801_1195 |
| Author(s) : Chahinian H , Fantini J , Garmy N , Manco G , Sarda L |
| Ref : Biochimica & Biophysica Acta , 1801 :1195 , 2010 |
| Abstract : Chahinian_2010_Biochim.Biophys.Acta_1801_1195 |
| ESTHER : Chahinian_2010_Biochim.Biophys.Acta_1801_1195 |
| PubMedSearch : Chahinian_2010_Biochim.Biophys.Acta_1801_1195 |
| PubMedID: 20655391 |
| Title : Activation of bacterial thermoalkalophilic lipases is spurred by dramatic structural rearrangements - Carrasco-Lopez_2009_J.Biol.Chem_284_4365 |
| Author(s) : Carrasco-Lopez C , Godoy C , de Las Rivas B , Fernandez-Lorente G , Palomo JM , Guisan JM , Fernandez-Lafuente R , Martinez-Ripoll M , Hermoso JA |
| Ref : Journal of Biological Chemistry , 284 :4365 , 2009 |
| Abstract : Carrasco-Lopez_2009_J.Biol.Chem_284_4365 |
| ESTHER : Carrasco-Lopez_2009_J.Biol.Chem_284_4365 |
| PubMedSearch : Carrasco-Lopez_2009_J.Biol.Chem_284_4365 |
| PubMedID: 19056729 |
| Gene_locus related to this paper: bactc-lipas |
| Title : Understanding promiscuous amidase activity of an esterase from Bacillus subtilis - |
| Author(s) : Kourist R , Bartsch S , Fransson L , Hult K , Bornscheuer UT |
| Ref : Chembiochem , 9 :67 , 2008 |
| PubMedID: 18022973 |
| Gene_locus related to this paper: bacsu-pnbae , canar-LipB |
| Title : Enzyme genomics: Application of general enzymatic screens to discover new enzymes - Kuznetsova_2005_FEMS.Microbiol.Rev_29_263 |
| Author(s) : Kuznetsova E , Proudfoot M , Sanders SA , Reinking J , Savchenko A , Arrowsmith CH , Edwards AM , Yakunin AF |
| Ref : FEMS Microbiology Reviews , 29 :263 , 2005 |
| Abstract : Kuznetsova_2005_FEMS.Microbiol.Rev_29_263 |
| ESTHER : Kuznetsova_2005_FEMS.Microbiol.Rev_29_263 |
| PubMedSearch : Kuznetsova_2005_FEMS.Microbiol.Rev_29_263 |
| PubMedID: 15808744 |
| Gene_locus related to this paper: ecoli-yafa , ecoli-ybff , ecoli-ycjy , ecoli-yeiG , ecoli-YFBB , ecoli-yjfp , ecoli-ypfh , ecoli-yqia , ecoli-yuar |
| Title : Molecular cloning and characterization of two thermostable carboxyl esterases from Geobacillus stearothermophilus - Ewis_2004_Gene_329_187 |
| Author(s) : Ewis HE , Abdelal AT , Lu CD |
| Ref : Gene , 329 :187 , 2004 |
| Abstract : Ewis_2004_Gene_329_187 |
| ESTHER : Ewis_2004_Gene_329_187 |
| PubMedSearch : Ewis_2004_Gene_329_187 |
| PubMedID: 15033540 |
| Gene_locus related to this paper: geost-est30 , geost-est50 |
| Title : Cloning and characterization of EstC from Burkholderia gladioli, a novel-type esterase related to plant enzymes - Reiter_2000_Appl.Microbiol.Biotechnol_54_778 |
| Author(s) : Reiter B , Glieder A , Talker D , Schwab H |
| Ref : Applied Microbiology & Biotechnology , 54 :778 , 2000 |
| Abstract : Reiter_2000_Appl.Microbiol.Biotechnol_54_778 |
| ESTHER : Reiter_2000_Appl.Microbiol.Biotechnol_54_778 |
| PubMedSearch : Reiter_2000_Appl.Microbiol.Biotechnol_54_778 |
| PubMedID: 11152069 |
| Gene_locus related to this paper: burgl-EstC |
| Title : Purification and partial characterization of a novel thermophilic carboxylesterase with high mesophilic specific activity - Wood_1995_Enzyme.Microb.Technol_17_816 |
| Author(s) : Wood AN , Fernandez-Lafuente R , Cowan DA |
| Ref : Enzyme Microb Technol , 17 :816 , 1995 |
| Abstract : Wood_1995_Enzyme.Microb.Technol_17_816 |
| ESTHER : Wood_1995_Enzyme.Microb.Technol_17_816 |
| PubMedSearch : Wood_1995_Enzyme.Microb.Technol_17_816 |
| PubMedID: 7576531 |
| Gene_locus related to this paper: geost-est50 |
| Title : Purification and properties of an Arthrobacter oxydans P52 carbamate hydrolase specific for the herbicide phenmedipham and nucleotide sequence of the corresponding gene - Pohlenz_1992_J.Bacteriol_174_6600 |
| Author(s) : Pohlenz HD , Boidol W , Schuttke I , Streber WR |
| Ref : Journal of Bacteriology , 174 :6600 , 1992 |
| Abstract : Pohlenz_1992_J.Bacteriol_174_6600 |
| ESTHER : Pohlenz_1992_J.Bacteriol_174_6600 |
| PubMedSearch : Pohlenz_1992_J.Bacteriol_174_6600 |
| PubMedID: 1400211 |
| Gene_locus related to this paper: artox-phhyd |