HomeoSplitter
HomeoSplitter: a software for disentangling homeologous contigs in allo-tetraploid assembly.
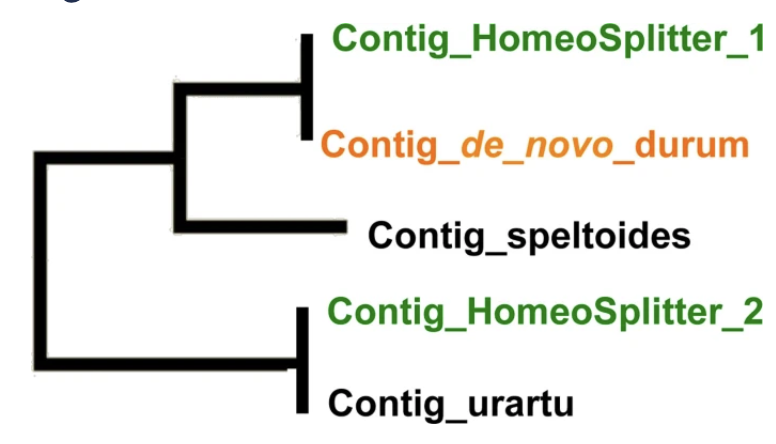
Using Next Generation Sequencing, SNP discovery is relatively easy on diploid species and still hampered in polyploid species by the confusion due to homeology. We develop HomeoSplitter; a fast and effective solution to split original contigs obtained by RNAseq into two homeologous sequences. It uses the differential expression of the two homeologous genes in the RNA. We verify that the new sequences are closer to the diploid progenitors of the allopolyploid species than the original contig. By remapping original reads on these new sequences, we also verify that the number of valuable detected SNPs has significantly increased.
HomeoSplitter is a fast and effective solution to disentangle homeologous sequences based on a maximum likelihood optimization. On a benchmark set of 2,505 clusters containing homologous sequences of urartu, speltoides and durum, HomeoSplitter was efficient to build sequences closer to the diploid references and increased the number of valuable SNPs from 188 out of 1,360 SNPs detected when mapping the reads on the de novo durum assembly to 762 out of 1,620 SNPs when mapping on HomeoSplitter contigs.
HomeoSplitter provides a practical solution to the complex problem of disentangling homeologous transcripts in allo-tetraploids, which further allows an improved SNP detection.