CHEMBL1818384
Also targets the inner membrane transporter MmpL3 of Mycobacterium tuberculosis with which it has been cristallized
General
Type : Adamantyl,Urea derivative
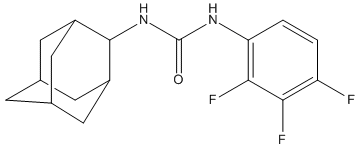
Chemical_Nomenclature : 1-(2-adamantyl)-3-(2,3,4-trifluorophenyl)urea
Canonical SMILES : C1C2CC3CC1CC(C2)C3NC(=O)NC4=C(C(=C(C=C4)F)F)F
InChI : InChI=1S\/C17H19F3N2O\/c18-12-1-2-13(15(20)14(12)19)21-17(23)22-16-10-4-8-3-9(6-10)7-11(16)5-8\/h1-2,8-11,16H,3-7H2,(H2,21,22,23)
InChIKey : FRRHMLGKNPFRKT-UHFFFAOYSA-N
Other name(s) :
MW : 324.35
Formula : C17H19F3N2O
CAS_number :
PubChem : 3754047
UniChem : FRRHMLGKNPFRKT-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
Families : CHEMBL1818384 ligand of proteins in family: Epoxide_hydrolase
Stucture :
Protein : human-EPHX2 || myctu-ephB || myctu-ephE
References (4)
| Title : Design, synthesis and anti-tuberculosis activity of 1-adamantyl-3-heteroaryl ureas with improved in vitro pharmacokinetic properties - North_2013_Bioorg.Med.Chem_21_2587 |
| Author(s) : North EJ , Scherman MS , Bruhn DF , Scarborough JS , Maddox MM , Jones V , Grzegorzewicz A , Yang L , Hess T , Morisseau C , Jackson M , McNeil MR , Lee RE |
| Ref : Bioorganic & Medicinal Chemistry , 21 :2587 , 2013 |
| Abstract : North_2013_Bioorg.Med.Chem_21_2587 |
| ESTHER : North_2013_Bioorg.Med.Chem_21_2587 |
| PubMedSearch : North_2013_Bioorg.Med.Chem_21_2587 |
| PubMedID: 23498915 |
| Title : Screening a library of 1600 adamantyl ureas for anti-Mycobacterium tuberculosis activity in vitro and for better physical chemical properties for bioavailability - Scherman_2012_Bioorg.Med.Chem_20_3255 |
| Author(s) : Scherman MS , North EJ , Jones V , Hess TN , Grzegorzewicz AE , Kasagami T , Kim IH , Merzlikin O , Lenaerts AJ , Lee RE , Jackson M , Morisseau C , McNeil MR |
| Ref : Bioorganic & Medicinal Chemistry , 20 :3255 , 2012 |
| Abstract : Scherman_2012_Bioorg.Med.Chem_20_3255 |
| ESTHER : Scherman_2012_Bioorg.Med.Chem_20_3255 |
| PubMedSearch : Scherman_2012_Bioorg.Med.Chem_20_3255 |
| PubMedID: 22522007 |
| Title : Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane - Grzegorzewicz_2012_Nat.Chem.Biol_8_334 |
| Author(s) : Grzegorzewicz AE , Pham H , Gundi VA , Scherman MS , North EJ , Hess T , Jones V , Gruppo V , Born SE , Kordulakova J , Chavadi SS , Morisseau C , Lenaerts AJ , Lee RE , McNeil MR , Jackson M |
| Ref : Nat Chemical Biology , 8 :334 , 2012 |
| Abstract : Grzegorzewicz_2012_Nat.Chem.Biol_8_334 |
| ESTHER : Grzegorzewicz_2012_Nat.Chem.Biol_8_334 |
| PubMedSearch : Grzegorzewicz_2012_Nat.Chem.Biol_8_334 |
| PubMedID: 22344175 |
| Title : The structure-activity relationship of urea derivatives as anti-tuberculosis agents - Brown_2011_Bioorg.Med.Chem_19_5585 |
| Author(s) : Brown JR , North EJ , Hurdle JG , Morisseau C , Scarborough JS , Sun D , Kordulakova J , Scherman MS , Jones V , Grzegorzewicz A , Crew RM , Jackson M , McNeil MR , Lee RE |
| Ref : Bioorganic & Medicinal Chemistry , 19 :5585 , 2011 |
| Abstract : Brown_2011_Bioorg.Med.Chem_19_5585 |
| ESTHER : Brown_2011_Bioorg.Med.Chem_19_5585 |
| PubMedSearch : Brown_2011_Bioorg.Med.Chem_19_5585 |
| PubMedID: 21840723 |