Cyclophostin
Natural organophosphate inhibitor of Acetylcholinesterase isolated from cultures of Streptomyces lavendulae strain NK901093. Compound very close to CGA-134-736 and CGA-134-735 described by Neumann 1987 Experientia 43 1235
General
Type : Organophosphate,Natural,Cyclic OP
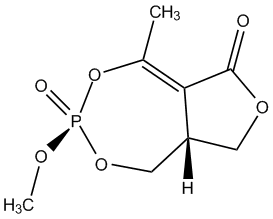
Chemical_Nomenclature : (3aR,6R)-3-methoxy-5-methyl-3-oxo-8,8a-dihydro-1H-furo[3,4-e][1,3,2]dioxaphosphepin-6-one
Canonical SMILES : CC1=C2C(COC2=O)COP(=O)(O1)OC
InChI : 1S\/C8H11O6P\/c1-5-7-6(3-12-8(7)9)4-13-15(10,11-2)14-5\/h6H,3-4H2,1-2H3\/t6-,15-\/m1\/s1
InChIKey : OMPQJMDGDAAXPE-NPMWZIQKSA-N
Other name(s) : CHEMBL603622,CHEBI:697555,TAN-1139
MW : 234.1
Formula : C8H11O6P
CAS_number : 144773-26-2
PubChem : 9578246
UniChem : OMPQJMDGDAAXPE-NPMWZIQKSA-N
IUPHAR :
Wikipedia :
Target
References (11)
| Title : Cyclophostin and Cyclipostins analogues, new promising molecules to treat mycobacterial-related diseases - Nguyen_2018_Int.J.Antimicrob.Agents_51_651 |
| Author(s) : Nguyen PC , Madani A , Santucci P , Martin BP , Paudel RR , Delattre S , Herrmann JL , Spilling CD , Kremer L , Canaan S , Cavalier JF |
| Ref : Int J Antimicrob Agents , 51 :651 , 2018 |
| Abstract : Nguyen_2018_Int.J.Antimicrob.Agents_51_651 |
| ESTHER : Nguyen_2018_Int.J.Antimicrob.Agents_51_651 |
| PubMedSearch : Nguyen_2018_Int.J.Antimicrob.Agents_51_651 |
| PubMedID: 29241819 |
| Title : Rat hormone sensitive lipase inhibition by cyclipostins and their analogs - Vasilieva_2015_Bioorg.Med.Chem_23_944 |
| Author(s) : Vasilieva E , Dutta S , Malla RK , Martin BP , Spilling CD , Dupureur CM |
| Ref : Bioorganic & Medicinal Chemistry , 23 :944 , 2015 |
| Abstract : Vasilieva_2015_Bioorg.Med.Chem_23_944 |
| ESTHER : Vasilieva_2015_Bioorg.Med.Chem_23_944 |
| PubMedSearch : Vasilieva_2015_Bioorg.Med.Chem_23_944 |
| PubMedID: 25678014 |
| Gene_locus related to this paper: ratno-hslip |
| Title : Synthesis and comparison of the biological activity of monocyclic phosphonate, difluorophosphonate and phosphate analogs of the natural AChE inhibitor cyclophostin - Martin_2015_Bioorg.Med.Chem_23_7529 |
| Author(s) : Martin BP , Vasilieva E , Dupureur CM , Spilling CD |
| Ref : Bioorganic & Medicinal Chemistry , 23 :7529 , 2015 |
| Abstract : Martin_2015_Bioorg.Med.Chem_23_7529 |
| ESTHER : Martin_2015_Bioorg.Med.Chem_23_7529 |
| PubMedSearch : Martin_2015_Bioorg.Med.Chem_23_7529 |
| PubMedID: 26585276 |
| Title : Enantioselective inhibition of microbial lipolytic enzymes by nonracemic monocyclic enolphosphonate analogues of cyclophostin - Point_2013_J.Med.Chem_56_4393 |
| Author(s) : Point V , Malla RK , Carriere F , Canaan S , Spilling CD , Cavalier JF |
| Ref : Journal of Medicinal Chemistry , 56 :4393 , 2013 |
| Abstract : Point_2013_J.Med.Chem_56_4393 |
| ESTHER : Point_2013_J.Med.Chem_56_4393 |
| PubMedSearch : Point_2013_J.Med.Chem_56_4393 |
| PubMedID: 23651298 |
| Title : Synthesis and kinetic evaluation of cyclophostin and cyclipostins phosphonate analogs as selective and potent inhibitors of microbial lipases - Point_2012_J.Med.Chem_55_10204 |
| Author(s) : Point V , Malla RK , Diomande S , Martin BP , Delorme V , Carriere F , Canaan S , Rath NP , Spilling CD , Cavalier JF |
| Ref : Journal of Medicinal Chemistry , 55 :10204 , 2012 |
| Abstract : Point_2012_J.Med.Chem_55_10204 |
| ESTHER : Point_2012_J.Med.Chem_55_10204 |
| PubMedSearch : Point_2012_J.Med.Chem_55_10204 |
| PubMedID: 23095026 |
| Title : The first total synthesis of (+\/-)-cyclophostin and (+\/-)-cyclipostin P: inhibitors of the serine hydrolases acetyl cholinesterase and hormone sensitive lipase - Malla_2011_Org.Lett_13_3094 |
| Author(s) : Malla RK , Bandyopadhyay S , Spilling CD , Dutta S , Dupureur CM |
| Ref : Org Lett , 13 :3094 , 2011 |
| Abstract : Malla_2011_Org.Lett_13_3094 |
| ESTHER : Malla_2011_Org.Lett_13_3094 |
| PubMedSearch : Malla_2011_Org.Lett_13_3094 |
| PubMedID: 21591624 |
| Title : Synthesis and kinetic analysis of some phosphonate analogs of cyclophostin as inhibitors of human acetylcholinesterase - Dutta_2010_Bioorg.Med.Chem_18_2265 |
| Author(s) : Dutta S , Malla RK , Bandyopadhyay S , Spilling CD , Dupureur CM |
| Ref : Bioorganic & Medicinal Chemistry , 18 :2265 , 2010 |
| Abstract : Dutta_2010_Bioorg.Med.Chem_18_2265 |
| ESTHER : Dutta_2010_Bioorg.Med.Chem_18_2265 |
| PubMedSearch : Dutta_2010_Bioorg.Med.Chem_18_2265 |
| PubMedID: 20189400 |
| Title : Synthesis and biological evaluation of a phosphonate analog of the natural acetyl cholinesterase inhibitor cyclophostin - Bandyopadhyay_2008_J.Org.Chem_73_8386 |
| Author(s) : Bandyopadhyay S , Dutta S , Spilling CD , Dupureur CM , Rath NP |
| Ref : J Org Chem , 73 :8386 , 2008 |
| Abstract : Bandyopadhyay_2008_J.Org.Chem_73_8386 |
| ESTHER : Bandyopadhyay_2008_J.Org.Chem_73_8386 |
| PubMedSearch : Bandyopadhyay_2008_J.Org.Chem_73_8386 |
| PubMedID: 18821801 |
| Title : Cyclipostins, novel hormone-sensitive lipase inhibitors from Streptomyces sp. DSM 13381. II. Isolation, structure elucidation and biological properties - Vertesy_2002_J.Antibiot.(Tokyo)_55_480 |
| Author(s) : Vertesy L , Beck B , Bronstrup M , Ehrlich K , Kurz M , Muller G , Schummer D , Seibert G |
| Ref : J Antibiot (Tokyo) , 55 :480 , 2002 |
| Abstract : Vertesy_2002_J.Antibiot.(Tokyo)_55_480 |
| ESTHER : Vertesy_2002_J.Antibiot.(Tokyo)_55_480 |
| PubMedSearch : Vertesy_2002_J.Antibiot.(Tokyo)_55_480 |
| PubMedID: 12139017 |
| Title : Conformational analysis of cyclophostin and designed analogs in comparison with the potent IP3 receptor agonist adenophostin A - Iwata_2001_Drug.Des.Discov_17_253 |
| Author(s) : Iwata Y , de Kort M , Challiss RA , van der Marel GA , van Boom JH , Miyamoto S |
| Ref : Drug Des Discov , 17 :253 , 2001 |
| Abstract : Iwata_2001_Drug.Des.Discov_17_253 |
| ESTHER : Iwata_2001_Drug.Des.Discov_17_253 |
| PubMedSearch : Iwata_2001_Drug.Des.Discov_17_253 |
| PubMedID: 11469755 |
| Title : Cyclophostin, acetylcholinesterase inhibitor from Streptomyces lavendulae - |
| Author(s) : Kurokawa T , Suzuki K , Hayaoka T , Nakagawa T , Izawa T , Kobayashi M , Harada N |
| Ref : J Antibiot (Tokyo) , 46 :1315 , 1993 |
| PubMedID: 8407597 |