Zifrosilone
General
Type : Halo ketone,Transition state analogue,Trifluoro
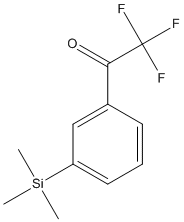
Chemical_Nomenclature : 2,2,2-Trifluoro-3'-(trimethylsilyl)acetophenone
Canonical SMILES : C[Si](C)(C)C1=CC=CC(=C1)C(=O)C(F)(F)F
InChI : InChI=1S\/C11H13F3OSi\/c1-16(2,3)9-6-4-5-8(7-9)10(15)11(12,13)14\/h4-7H,1-3H3
InChIKey : GAPOASFZXBWUGS-UHFFFAOYSA-N
Other name(s) : MDL-73745,MDL 73,745,MDL 73745,BRN 7373588,2,2,2-Trifluoro-3'-(trimethylsilyl)acetophenone,2,2,2-Trifluoro-1-(3-(trimethylsilyl)phenyl)ethanone,Ethanone, 2,2,2-trifluoro-1-(3-(trimethylsilyl)phenyl)-,2,2,2-trifluoro-1-(3-trimethylsilylphenyl) ethanone
MW : 246.301
Formula : C11H13F3OSi
CAS_number : 132236-18-1
PubChem : 60811
UniChem : GAPOASFZXBWUGS-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
References (7)
| Title : Drug and pro-drug substrates and pseudo-substrates of human butyrylcholinesterase - Masson_2023_Biochem.Pharmacol_218_115910 |
| Author(s) : Masson P , Shaihutdinova Z , Lockridge O |
| Ref : Biochemical Pharmacology , 218 :115910 , 2023 |
| Abstract : Masson_2023_Biochem.Pharmacol_218_115910 |
| ESTHER : Masson_2023_Biochem.Pharmacol_218_115910 |
| PubMedSearch : Masson_2023_Biochem.Pharmacol_218_115910 |
| PubMedID: 37972875 |
| Title : 1-(3-Tert-Butylphenyl)-2,2,2-Trifluoroethanone as a Potent Transition-State Analogue Slow-Binding Inhibitor of Human Acetylcholinesterase: Kinetic, MD and QM\/MM Studies - Zueva_2020_Biomolecules_10_1608 |
| Author(s) : Zueva IV , Lushchekina SV , Pottie IR , Darvesh S , Masson P |
| Ref : Biomolecules , 10 : , 2020 |
| Abstract : Zueva_2020_Biomolecules_10_1608 |
| ESTHER : Zueva_2020_Biomolecules_10_1608 |
| PubMedSearch : Zueva_2020_Biomolecules_10_1608 |
| PubMedID: 33260981 |
| Title : Effect of MDL 73,745 on acetylcholine and biogenic amine levels in rat cortex - Zhu_1995_Eur.J.Pharmacol_276_93 |
| Author(s) : Zhu XD , Giacobini E , Hornsperger JM |
| Ref : European Journal of Pharmacology , 276 :93 , 1995 |
| Abstract : Zhu_1995_Eur.J.Pharmacol_276_93 |
| ESTHER : Zhu_1995_Eur.J.Pharmacol_276_93 |
| PubMedSearch : Zhu_1995_Eur.J.Pharmacol_276_93 |
| PubMedID: 7781701 |
| Title : Pharmacokinetics and pharmacodynamics of the acetylcholinesterase inhibitor 2,2,2-trifluoro-1-(3-trimethylsilylphenyl) ethanone in dog. Potential for transdermal patch delivery - Dow_1995_Arzneimittelforschung_45_1245 |
| Author(s) : Dow J , Dulery BD , Hornsperger JM , Di Francesco GF , Keshary P , Haegele KD |
| Ref : Arzneimittelforschung , 45 :1245 , 1995 |
| Abstract : Dow_1995_Arzneimittelforschung_45_1245 |
| ESTHER : Dow_1995_Arzneimittelforschung_45_1245 |
| PubMedSearch : Dow_1995_Arzneimittelforschung_45_1245 |
| PubMedID: 8595078 |
| Title : Acetylcholinesterase inhibition by zifrosilone: pharmacokinetics and pharmacodynamics - Cutler_1995_Clin.Pharmacol.Ther_58_54 |
| Author(s) : Cutler NR , Seifert RD , Schleman MM , Sramek JJ , Szylleyko OJ , Howard DR , Barchowsky A , Wardle TS , Brass EP |
| Ref : Clinical Pharmacology & Therapeutics , 58 :54 , 1995 |
| Abstract : Cutler_1995_Clin.Pharmacol.Ther_58_54 |
| ESTHER : Cutler_1995_Clin.Pharmacol.Ther_58_54 |
| PubMedSearch : Cutler_1995_Clin.Pharmacol.Ther_58_54 |
| PubMedID: 7628183 |
| Title : Trimethylsilylated trifluoromethyl ketones, a novel class of acetylcholinesterase inhibitors: biochemical and pharmacological profile of MDL 73,745 - |
| Author(s) : Hornsperger JM , Collard JN , Heydt JG , Giacobini E , Funes S , Dow J , Schirlin D |
| Ref : Biochemical Society Transactions , 22 :758 , 1994 |
| PubMedID: 7821680 |
| Title : Determination of an acetylcholinesterase inhibitor, MDL 73745, in dog plasma and urine by combined gas chromatography\/negative ion chemical ionization mass spectrometry - Dulery_1993_Biol.Mass.Spectrom_22_377 |
| Author(s) : Dulery BD , Le Henanf N , Dow J , Haegele KD |
| Ref : Biol Mass Spectrom , 22 :377 , 1993 |
| Abstract : Dulery_1993_Biol.Mass.Spectrom_22_377 |
| ESTHER : Dulery_1993_Biol.Mass.Spectrom_22_377 |
| PubMedSearch : Dulery_1993_Biol.Mass.Spectrom_22_377 |
| PubMedID: 8357853 |