Flutamide
General
Type : Trifluoro
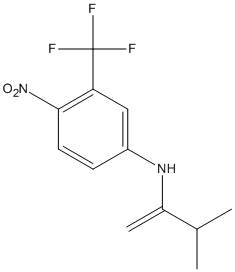
Chemical_Nomenclature : 2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]propanamide
Canonical SMILES : CC(C)C(=O)NC1=CC(=C(C=C1)[N+](=O)[O-])C(F)(F)F
InChI : InChI=1S\/C11H11F3N2O3\/c1-6(2)10(17)15-7-3-4-9(16(18)19)8(5-7)11(12,13)14\/h3-6H,1-2H3,(H,15,17)
InChIKey : MKXKFYHWDHIYRV-UHFFFAOYSA-N
Other name(s) : Eulexin, Niftolide, Niftholide
Target
Families : Arylacetamide_deacetylase
References (4)
| Title : Contributions of arylacetamide deacetylase and carboxylesterase 2 to flutamide hydrolysis in human liver - Kobayashi_2012_Drug.Metab.Dispos_40_1080 |
| Author(s) : Kobayashi Y , Fukami T , Shimizu M , Nakajima M , Yokoi T |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 40 :1080 , 2012 |
| Abstract : Kobayashi_2012_Drug.Metab.Dispos_40_1080 |
| ESTHER : Kobayashi_2012_Drug.Metab.Dispos_40_1080 |
| PubMedSearch : Kobayashi_2012_Drug.Metab.Dispos_40_1080 |
| PubMedID: 22446520 |
| Title : Human arylacetamide deacetylase is a principal enzyme in flutamide hydrolysis - Watanabe_2009_Drug.Metab.Dispos_37_1513 |
| Author(s) : Watanabe A , Fukami T , Nakajima M , Takamiya M , Aoki Y , Yokoi T |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 37 :1513 , 2009 |
| Abstract : Watanabe_2009_Drug.Metab.Dispos_37_1513 |
| ESTHER : Watanabe_2009_Drug.Metab.Dispos_37_1513 |
| PubMedSearch : Watanabe_2009_Drug.Metab.Dispos_37_1513 |
| PubMedID: 19339378 |
| Gene_locus related to this paper: human-AADAC |
| Title : Detection of a new N-oxidized metabolite of flutamide, N-[4-nitro-3-(trifluoromethyl)phenyl]hydroxylamine, in human liver microsomes and urine of prostate cancer patients - Goda_2006_Drug.Metab.Dispos_34_828 |
| Author(s) : Goda R , Nagai D , Akiyama Y , Nishikawa K , Ikemoto I , Aizawa Y , Nagata K , Yamazoe Y |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 34 :828 , 2006 |
| Abstract : Goda_2006_Drug.Metab.Dispos_34_828 |
| ESTHER : Goda_2006_Drug.Metab.Dispos_34_828 |
| PubMedSearch : Goda_2006_Drug.Metab.Dispos_34_828 |
| PubMedID: 16507648 |
| Title : [Changes in liver function induced by flutamide in patients with prostate cancer (studies in patients treated with total androgen blockage)] - Ikemoto_2000_Nihon.Hinyokika.Gakkai.Zasshi_91_556 |
| Author(s) : Ikemoto I , Ohishi Y , Yamazaki H , Wada T , Aizawa Y |
| Ref : Nihon Hinyokika Gakkai Zasshi , 91 :556 , 2000 |
| Abstract : Ikemoto_2000_Nihon.Hinyokika.Gakkai.Zasshi_91_556 |
| ESTHER : Ikemoto_2000_Nihon.Hinyokika.Gakkai.Zasshi_91_556 |
| PubMedSearch : Ikemoto_2000_Nihon.Hinyokika.Gakkai.Zasshi_91_556 |
| PubMedID: 10897581 |