Neostigmine~Prostigmine
A cholinesterase inhibitor used in the treatment of myasthenia gravis and to reverse the effects of muscle relaxants such as gallamine and tubocurarine. Neostigmine, unlike PHYSOSTIGMINE- ESERINE, does not cross the blood-brain barrier. Parasympathomimetic as well as Neostigmine Methyl Sulfate. Used to reverse action of neuromuscular blockers and snake venoms. There is also Nor-physostigmine(...-dimethyl aminoformate).Neostigmine sulfoxymethane (Juvastigmin,Kirkstigmine, Neostigmeth, Normastigmin, Polstigmine, Synstigmine, Syntostigmin, Hodostin, Proserin Hoffman-La-Roche prostigmine was marketed in the early 1930s for the treatment of glaucoma, myasthenia gravis, to increase gut and bladder functions.
General
Type : Carbamate,Trimethylammonium
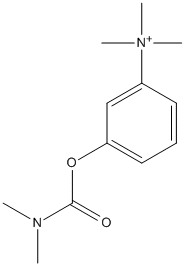
Chemical_Nomenclature : [3-(dimethylcarbamoyloxy)phenyl]-trimethylazanium
Canonical SMILES : CN(C)C(=O)OC1=CC=CC(=C1)[N+](C)(C)C || CN(C)C(=O)OC1=CC=CC(=C1)[N+](C)(C)C.COS(=O)(=O)O
InChI : InChI=1S\/C12H19N2O2\/c1-13(2)12(15)16-11-8-6-7-10(9-11)14(3,4)5\/h6-9H,1-5H3\/q+1 || InChI=1S\/C12H19N2O2.CH4O4S\/c1-13(2)12(15)16-11-8-6-7-10(9-11)14(3,4)5\;1-5-6(2,3)4\/h6-9H,1-5H3\;1H3,(H,2,3,4)\/q+1\;
InChIKey : ALWKGYPQUAPLQC-UHFFFAOYSA-N || OSZNNLWOYWAHSS-UHFFFAOYSA-N
Other name(s) : (m-hydroxyphenyl) trimethylammonium bromide dimethyl carbamate,(3-dimethylcarbamoxyphenyl)trimethylammonium bromide,Kirkstigmine,Neostigmeth,Normastigmin,Polstigmine,Synstigmine,Syntostigmin,Hodostin,Proserin,Neo proserine,neo-Proserin,Eustigmin,Neoserine,Proserine,Stigmanol,Stigmosan,Prostigmin,Prostigmine,Synstigmin,Neostigmine methosulfate,Neostigmine,Philostigmine,Eustigmine
MW : 303.21
Formula : C12H19BrN2O2
CAS_number : 114-80-7 || 59-99-4
UniChem : ALWKGYPQUAPLQC-UHFFFAOYSA-N || OSZNNLWOYWAHSS-UHFFFAOYSA-N
IUPHAR : 8993
Wikipedia : Neostigmine
Target
References (66)
| Title : Recycling of acetylcholine following impulse transmission in rat muscle revealed in the presence of neostigmine - Uramoto_1998_Clin.Exp.Pharmacol.Physiol_25_44 |
| Author(s) : Uramoto I , Miyamoto K , Watanabe K , Totsuka T |
| Ref : Clinical & Experimental Pharmacology & Physiology , 25 :44 , 1998 |
| Abstract : Uramoto_1998_Clin.Exp.Pharmacol.Physiol_25_44 |
| ESTHER : Uramoto_1998_Clin.Exp.Pharmacol.Physiol_25_44 |
| PubMedSearch : Uramoto_1998_Clin.Exp.Pharmacol.Physiol_25_44 |
| PubMedID: 9493558 |
| Title : Neostigmine competitively inhibited nicotinic acetylcholine receptors in sympathetic neurons - Zheng_1998_Life.Sci_62_1171 |
| Author(s) : Zheng JQ , He XP , Yang AZ , Liu CG |
| Ref : Life Sciences , 62 :1171 , 1998 |
| Abstract : Zheng_1998_Life.Sci_62_1171 |
| ESTHER : Zheng_1998_Life.Sci_62_1171 |
| PubMedSearch : Zheng_1998_Life.Sci_62_1171 |
| PubMedID: 9519798 |
| Title : Does neostigmine have a deleterious effect on the resistance of colonic anastomoses? - Garcia-Olmo_1998_Eur.J.Anaesthesiol_15_38 |
| Author(s) : Garcia-Olmo DC , Garcia-Rivas M , Garcia-Olmo D |
| Ref : European Journal of Anaesthesiology , 15 :38 , 1998 |
| Abstract : Garcia-Olmo_1998_Eur.J.Anaesthesiol_15_38 |
| ESTHER : Garcia-Olmo_1998_Eur.J.Anaesthesiol_15_38 |
| PubMedSearch : Garcia-Olmo_1998_Eur.J.Anaesthesiol_15_38 |
| PubMedID: 9522139 |
| Title : Effects of oral clonidine on heart rate changes after neostigmine- atropine administration - Kimura_1998_Anesthesiology_88_1507 |
| Author(s) : Kimura T , Tanaka M , Nishikawa T |
| Ref : Anesthesiology , 88 :1507 , 1998 |
| Abstract : Kimura_1998_Anesthesiology_88_1507 |
| ESTHER : Kimura_1998_Anesthesiology_88_1507 |
| PubMedSearch : Kimura_1998_Anesthesiology_88_1507 |
| PubMedID: 9637644 |
| Title : The efficacy of intrathecal neostigmine, intrathecal morphine, and their combination for post-cesarean section analgesia - Chung_1998_Anesth.Analg_87_341 |
| Author(s) : Chung CJ , Kim JS , Park HS , Chin YJ |
| Ref : Anesthesia & Analgesia , 87 :341 , 1998 |
| Abstract : Chung_1998_Anesth.Analg_87_341 |
| ESTHER : Chung_1998_Anesth.Analg_87_341 |
| PubMedSearch : Chung_1998_Anesth.Analg_87_341 |
| PubMedID: 9706928 |
| Title : Intrathecal neostigmine, but not sympathectomy, relieves mechanical allodynia in a rat model of neuropathic pain - Lavand'homme_1998_Anesthesiology_89_493 |
| Author(s) : Lavand'homme P , Pan HL , Eisenach JC |
| Ref : Anesthesiology , 89 :493 , 1998 |
| Abstract : Lavand'homme_1998_Anesthesiology_89_493 |
| ESTHER : Lavand'homme_1998_Anesthesiology_89_493 |
| PubMedSearch : Lavand'homme_1998_Anesthesiology_89_493 |
| PubMedID: 9710409 |
| Title : Effects of four anticholinesterase-anticholinergic combinations and tracheal extubation on QTc interval of the ECG, heart rate and arterial pressure - Saarnivaara_1998_Acta.Anaesthesiol.Scand_42_460 |
| Author(s) : Saarnivaara L , Simola M |
| Ref : Acta Anaesthesiologica Scandinavica , 42 :460 , 1998 |
| Abstract : Saarnivaara_1998_Acta.Anaesthesiol.Scand_42_460 |
| ESTHER : Saarnivaara_1998_Acta.Anaesthesiol.Scand_42_460 |
| PubMedSearch : Saarnivaara_1998_Acta.Anaesthesiol.Scand_42_460 |
| PubMedID: 9563867 |
| Title : Postoperative analgesia by intra-articular neostigmine in patients undergoing knee arthroscopy - Yang_1998_Anesthesiology_88_334 |
| Author(s) : Yang LC , Chen LM , Wang CJ , Buerkle H |
| Ref : Anesthesiology , 88 :334 , 1998 |
| Abstract : Yang_1998_Anesthesiology_88_334 |
| ESTHER : Yang_1998_Anesthesiology_88_334 |
| PubMedSearch : Yang_1998_Anesthesiology_88_334 |
| PubMedID: 9477052 |
| Title : Effects of neostigmine and atropine on basal and handling-induced acetylcholine output from ventral hippocampus - Moor_1998_Neurosci_82_819 |
| Author(s) : Moor E , Schirm E , Jacso J , Westerink BH |
| Ref : Neuroscience , 82 :819 , 1998 |
| Abstract : Moor_1998_Neurosci_82_819 |
| ESTHER : Moor_1998_Neurosci_82_819 |
| PubMedSearch : Moor_1998_Neurosci_82_819 |
| PubMedID: 9483538 |
| Title : Dopaminergic regulation of striatal acetylcholine release: the critical role of acetylcholinesterase inhibition - Acquas_1998_J.Neurochem_70_1088 |
| Author(s) : Acquas E , Fibiger HC |
| Ref : Journal of Neurochemistry , 70 :1088 , 1998 |
| Abstract : Acquas_1998_J.Neurochem_70_1088 |
| ESTHER : Acquas_1998_J.Neurochem_70_1088 |
| PubMedSearch : Acquas_1998_J.Neurochem_70_1088 |
| PubMedID: 9489729 |
| Title : Effects of intrathecal neostigmine, bupivacaine, and their combination on sympathetic nerve activity in rats - Pan_1998_Anesthesiology_88_481 |
| Author(s) : Pan HL , Song HK , Eisenach JC |
| Ref : Anesthesiology , 88 :481 , 1998 |
| Abstract : Pan_1998_Anesthesiology_88_481 |
| ESTHER : Pan_1998_Anesthesiology_88_481 |
| PubMedSearch : Pan_1998_Anesthesiology_88_481 |
| PubMedID: 9477069 |
| Title : Central and peripheral analgesia mediated by the acetylcholinesterase- inhibitor neostigmine in the rat inflamed knee joint model - Buerkle_1998_Anesth.Analg_86_1027 |
| Author(s) : Buerkle H , Boschin M , Marcus MA , Brodner G , Wusten R , Van Aken H |
| Ref : Anesthesia & Analgesia , 86 :1027 , 1998 |
| Abstract : Buerkle_1998_Anesth.Analg_86_1027 |
| ESTHER : Buerkle_1998_Anesth.Analg_86_1027 |
| PubMedSearch : Buerkle_1998_Anesth.Analg_86_1027 |
| PubMedID: 9585291 |
| Title : Effects of local cholinesterase inhibition on acetylcholine release assessed simultaneously in prefrontal and frontoparietal cortex - Himmelheber_1998_Neurosci_86_949 |
| Author(s) : Himmelheber AM , Fadel J , Sarter M , Bruno JP |
| Ref : Neuroscience , 86 :949 , 1998 |
| Abstract : Himmelheber_1998_Neurosci_86_949 |
| ESTHER : Himmelheber_1998_Neurosci_86_949 |
| PubMedSearch : Himmelheber_1998_Neurosci_86_949 |
| PubMedID: 9692730 |
| Title : Inhibition of cholinesterase-associated aryl acylamidase activity by anticholinesterase agents: focus on drugs potentially effective in Alzheimer's disease - Costagli_1998_Biochem.Pharmacol_55_1733 |
| Author(s) : Costagli C , Galli A |
| Ref : Biochemical Pharmacology , 55 :1733 , 1998 |
| Abstract : Costagli_1998_Biochem.Pharmacol_55_1733 |
| ESTHER : Costagli_1998_Biochem.Pharmacol_55_1733 |
| PubMedSearch : Costagli_1998_Biochem.Pharmacol_55_1733 |
| PubMedID: 9634011 |
| Title : High dose neostigmine treatment of malignant sinus tachycardia - Schultheis_1997_Pacing.Clin.Electrophysiol_20_1369 |
| Author(s) : Schultheis LW , Balser JR , Calkins H , Robertson S , Weiss JL , Sussman M , Stuart RS |
| Ref : Pacing Clin Electrophysiol , 20 :1369 , 1997 |
| Abstract : Schultheis_1997_Pacing.Clin.Electrophysiol_20_1369 |
| ESTHER : Schultheis_1997_Pacing.Clin.Electrophysiol_20_1369 |
| PubMedSearch : Schultheis_1997_Pacing.Clin.Electrophysiol_20_1369 |
| PubMedID: 9170142 |
| Title : Intrathecal neostigmine for post-cesarean section analgesia: dose response - Krukowski_1997_Anesth.Analg_84_1269 |
| Author(s) : Krukowski JA , Hood DD , Eisenach JC , Mallak KA , Parker RL |
| Ref : Anesthesia & Analgesia , 84 :1269 , 1997 |
| Abstract : Krukowski_1997_Anesth.Analg_84_1269 |
| ESTHER : Krukowski_1997_Anesth.Analg_84_1269 |
| PubMedSearch : Krukowski_1997_Anesth.Analg_84_1269 |
| PubMedID: 9174305 |
| Title : Factors affecting neostigmine reversal of vecuronium block during sevoflurane anaesthesia - Morita_1997_Anaesthesia_52_538 |
| Author(s) : Morita T , Kurosaki D , Tsukagoshi H , Shimada H , Sato H , Goto F |
| Ref : Anaesthesia , 52 :538 , 1997 |
| Abstract : Morita_1997_Anaesthesia_52_538 |
| ESTHER : Morita_1997_Anaesthesia_52_538 |
| PubMedSearch : Morita_1997_Anaesthesia_52_538 |
| PubMedID: 9203879 |
| Title : Postoperative analgesic effect of intrathecal neostigmine and its influence on spinal anaesthesia - Klamt_1997_Anaesthesia_52_547 |
| Author(s) : Klamt JG , Slullitel A , Garcia IV , Prado WA |
| Ref : Anaesthesia , 52 :547 , 1997 |
| Abstract : Klamt_1997_Anaesthesia_52_547 |
| ESTHER : Klamt_1997_Anaesthesia_52_547 |
| PubMedSearch : Klamt_1997_Anaesthesia_52_547 |
| PubMedID: 9203881 |
| Title : Antinociceptive effect of intrathecal neostigmine evaluated in rats by two different pain models - Prado_1997_Braz.J.Med.Biol.Res_30_1225 |
| Author(s) : Prado WA , Goncalves AS |
| Ref : Brazilian Journal of Medical & Biological Research , 30 :1225 , 1997 |
| Abstract : Prado_1997_Braz.J.Med.Biol.Res_30_1225 |
| ESTHER : Prado_1997_Braz.J.Med.Biol.Res_30_1225 |
| PubMedSearch : Prado_1997_Braz.J.Med.Biol.Res_30_1225 |
| PubMedID: 9496442 |
| Title : Effect of glyceryl trinitrate on the sphincter of Oddi spasm evoked by prostigmine-morphine administration - Velosy_1997_Eur.J.Gastroenterol.Hepatol_9_1109 |
| Author(s) : Velosy B , Madacsy L , Lonovics J , Csernay L |
| Ref : European Journal of Gastroenterology & Hepatology , 9 :1109 , 1997 |
| Abstract : Velosy_1997_Eur.J.Gastroenterol.Hepatol_9_1109 |
| ESTHER : Velosy_1997_Eur.J.Gastroenterol.Hepatol_9_1109 |
| PubMedSearch : Velosy_1997_Eur.J.Gastroenterol.Hepatol_9_1109 |
| PubMedID: 9431903 |
| Title : Cholinesterase activity in human pulmonary arteries and veins - Walch_1997_Br.J.Pharmacol_121_986 |
| Author(s) : Walch L , Taisne C , Gascard JP , Nashashibi N , Brink C , Norel X |
| Ref : British Journal of Pharmacology , 121 :986 , 1997 |
| Abstract : Walch_1997_Br.J.Pharmacol_121_986 |
| ESTHER : Walch_1997_Br.J.Pharmacol_121_986 |
| PubMedSearch : Walch_1997_Br.J.Pharmacol_121_986 |
| PubMedID: 9222557 |
| Title : Mary B. Walker, M.D. and the pioneering use of prostigmin to treat myasthenia gravis - Keeney_1997_Doc.Ophthalmol_93_125 |
| Author(s) : Keeney AH , Keeney VT |
| Ref : Doc Ophthalmol , 93 :125 , 1997 |
| Abstract : Keeney_1997_Doc.Ophthalmol_93_125 |
| ESTHER : Keeney_1997_Doc.Ophthalmol_93_125 |
| PubMedSearch : Keeney_1997_Doc.Ophthalmol_93_125 |
| PubMedID: 9476610 |
| Title : Studies on the safety of glucose and paraben-containing neostigmine for intrathecal administration - Gurun_1997_Anesth.Analg_85_317 |
| Author(s) : Gurun MS , Leinbach R , Moore L , Lee CS , Owen MD , Eisenach JC |
| Ref : Anesthesia & Analgesia , 85 :317 , 1997 |
| Abstract : Gurun_1997_Anesth.Analg_85_317 |
| ESTHER : Gurun_1997_Anesth.Analg_85_317 |
| PubMedSearch : Gurun_1997_Anesth.Analg_85_317 |
| PubMedID: 9249107 |
| Title : Neostigmine but not edrophonium prolongs the action of mivacurium - Symington_1996_Can.J.Anaesth_43_1220 |
| Author(s) : Symington MJ , Mirakhur RK , Kumar N |
| Ref : Canadian Journal of Anaesthesia , 43 :1220 , 1996 |
| Abstract : Symington_1996_Can.J.Anaesth_43_1220 |
| ESTHER : Symington_1996_Can.J.Anaesth_43_1220 |
| PubMedSearch : Symington_1996_Can.J.Anaesth_43_1220 |
| PubMedID: 8955970 |
| Title : Neostigmine reversal of vecuronium neuromuscular block and the influence of renal failure - Dhonneur_1996_Anesth.Analg_82_134 |
| Author(s) : Dhonneur G , Rebaine C , Slavov V , Ruggier R , De Chaubry V , Duvaldestin P |
| Ref : Anesthesia & Analgesia , 82 :134 , 1996 |
| Abstract : Dhonneur_1996_Anesth.Analg_82_134 |
| ESTHER : Dhonneur_1996_Anesth.Analg_82_134 |
| PubMedSearch : Dhonneur_1996_Anesth.Analg_82_134 |
| PubMedID: 8712389 |
| Title : Influence and relative sensitivities of 50-Hz and 100-Hz tetanic stimuli on subsequent tetanic fade ratios in patients receiving vecuronium - Baurain_1996_Anesth.Analg_82_139 |
| Author(s) : Baurain MJ , Hoton F , Dernovoi BS , d'Hollander AA |
| Ref : Anesthesia & Analgesia , 82 :139 , 1996 |
| Abstract : Baurain_1996_Anesth.Analg_82_139 |
| ESTHER : Baurain_1996_Anesth.Analg_82_139 |
| PubMedSearch : Baurain_1996_Anesth.Analg_82_139 |
| PubMedID: 8712390 |
| Title : Neostigmine for the treatment of neurotoxicity following envenomation by the Asiatic cobra - Gold_1996_Annals.of.Emergency.Medicine_28_87 |
| Author(s) : Gold BS |
| Ref : Annals of Emergency Medicine , 28 :87 , 1996 |
| Abstract : Gold_1996_Annals.of.Emergency.Medicine_28_87 |
| ESTHER : Gold_1996_Annals.of.Emergency.Medicine_28_87 |
| PubMedSearch : Gold_1996_Annals.of.Emergency.Medicine_28_87 |
| PubMedID: 8669746 |
| Title : Neostigmine decreases heart rate in heart transplant patients - Backman_1996_Can.J.Anaesth_43_373 |
| Author(s) : Backman SB , Fox GS , Stein RD , Ralley FE |
| Ref : Canadian Journal of Anaesthesia , 43 :373 , 1996 |
| Abstract : Backman_1996_Can.J.Anaesth_43_373 |
| ESTHER : Backman_1996_Can.J.Anaesth_43_373 |
| PubMedSearch : Backman_1996_Can.J.Anaesth_43_373 |
| PubMedID: 8697553 |
| Title : Dose responses for neostigmine and edrophonium as antagonists of mivacurium in adults and children - Bevan_1996_Anesthesiology_84_354 |
| Author(s) : Bevan JC , Tousignant C , Stephenson C , Blackman L , Reimer E , Smith MF , Bevan DR |
| Ref : Anesthesiology , 84 :354 , 1996 |
| Abstract : Bevan_1996_Anesthesiology_84_354 |
| ESTHER : Bevan_1996_Anesthesiology_84_354 |
| PubMedSearch : Bevan_1996_Anesthesiology_84_354 |
| PubMedID: 8602666 |
| Title : Neostigmine-induced bradycardia following recent vs remote cardiac transplantation in the same patient - Backman_1996_Can.J.Anaesth_43_394 |
| Author(s) : Backman SB , Stein RD , Ralley FE , Fox GS |
| Ref : Canadian Journal of Anaesthesia , 43 :394 , 1996 |
| Abstract : Backman_1996_Can.J.Anaesth_43_394 |
| ESTHER : Backman_1996_Can.J.Anaesth_43_394 |
| PubMedSearch : Backman_1996_Can.J.Anaesth_43_394 |
| PubMedID: 8697556 |
| Title : Anticholinesterase drugs stimulate phosphatidylinositol response in rat tracheal slices - Shibata_1996_Anesth.Analg_82_1211 |
| Author(s) : Shibata O , Kanairo M , Zhang S , Hasuo H , Morooka H , Fujie T , Sumikawa K |
| Ref : Anesthesia & Analgesia , 82 :1211 , 1996 |
| Abstract : Shibata_1996_Anesth.Analg_82_1211 |
| ESTHER : Shibata_1996_Anesth.Analg_82_1211 |
| PubMedSearch : Shibata_1996_Anesth.Analg_82_1211 |
| PubMedID: 8638793 |
| Title : Different properties of the bradycardia produced by neostigmine and edrophonium in the cat - Backman_1996_Can.J.Anaesth_43_731 |
| Author(s) : Backman SB , Stein RD , Blank DW , Collier B , Polosa C |
| Ref : Canadian Journal of Anaesthesia , 43 :731 , 1996 |
| Abstract : Backman_1996_Can.J.Anaesth_43_731 |
| ESTHER : Backman_1996_Can.J.Anaesth_43_731 |
| PubMedSearch : Backman_1996_Can.J.Anaesth_43_731 |
| PubMedID: 8807181 |
| Title : Neostigmine in the treatment of snake accidents caused by Micrurus frontalis: report of two cases (1) - Vital_1996_Rev.Inst.Med.Trop.Sao.Paulo_38_61 |
| Author(s) : Vital Brazil O , Vieira RJ |
| Ref : Revista do Instituto de Medicina Tropical de Sao Paulo , 38 :61 , 1996 |
| Abstract : Vital_1996_Rev.Inst.Med.Trop.Sao.Paulo_38_61 |
| ESTHER : Vital_1996_Rev.Inst.Med.Trop.Sao.Paulo_38_61 |
| PubMedSearch : Vital_1996_Rev.Inst.Med.Trop.Sao.Paulo_38_61 |
| PubMedID: 8762642 |
| Title : Interaction between intrathecal neostigmine and epidural clonidine in human volunteers - Hood_1996_Anesthesiology_85_315 |
| Author(s) : Hood DD , Mallak KA , Eisenach JC , Tong C |
| Ref : Anesthesiology , 85 :315 , 1996 |
| Abstract : Hood_1996_Anesthesiology_85_315 |
| ESTHER : Hood_1996_Anesthesiology_85_315 |
| PubMedSearch : Hood_1996_Anesthesiology_85_315 |
| PubMedID: 8712447 |
| Title : Do we need anti snake venom (ASV) for management of elapid ophitoxaemia - Bomb_1996_Journal.of.the.Association.of.Physicians.of.India_44_31 |
| Author(s) : Bomb BS , Roy S , Kumawat DC , Bharjatya M |
| Ref : Journal of the Association of Physicians of India , 44 :31 , 1996 |
| Abstract : Bomb_1996_Journal.of.the.Association.of.Physicians.of.India_44_31 |
| ESTHER : Bomb_1996_Journal.of.the.Association.of.Physicians.of.India_44_31 |
| PubMedSearch : Bomb_1996_Journal.of.the.Association.of.Physicians.of.India_44_31 |
| PubMedID: 8773091 |
| Title : Conditions to optimise the reversal action of neostigmine upon a vecuronium-induced neuromuscular block - Baurain_1996_Acta.Anaesthesiol.Scand_40_574 |
| Author(s) : Baurain MJ , Dernovoi BS , d'Hollander AA , Hennart DA , Cantraine FR |
| Ref : Acta Anaesthesiologica Scandinavica , 40 :574 , 1996 |
| Abstract : Baurain_1996_Acta.Anaesthesiol.Scand_40_574 |
| ESTHER : Baurain_1996_Acta.Anaesthesiol.Scand_40_574 |
| PubMedSearch : Baurain_1996_Acta.Anaesthesiol.Scand_40_574 |
| PubMedID: 8792887 |
| Title : Optimum time for neostigmine reversal of atracurium-induced neuromuscular blockade - Kirkegaard-Nielsen_1996_Can.J.Anaesth_43_932 |
| Author(s) : Kirkegaard-Nielsen H , Helbo-Hansen HS , Lindholm P , Severinsen IK , Pedersen HS , Jensen EW |
| Ref : Canadian Journal of Anaesthesia , 43 :932 , 1996 |
| Abstract : Kirkegaard-Nielsen_1996_Can.J.Anaesth_43_932 |
| ESTHER : Kirkegaard-Nielsen_1996_Can.J.Anaesth_43_932 |
| PubMedSearch : Kirkegaard-Nielsen_1996_Can.J.Anaesth_43_932 |
| PubMedID: 8874911 |
| Title : Additives in neuraxial anesthesia: opioids, alpha-2 adrenergic agonists, and neostigmine as a possible future drug for perioperative pain management - |
| Author(s) : Klimscha W , Chiari A , Lorber C , Krenn K , Dumitrescu R , Agnese T |
| Ref : Acta Anaesthesiologica Scandinavica Supplementum , 109 :176 , 1996 |
| PubMedID: 8901999 |
| Title : Is recovery of neuromuscular transmission complete after the use of neostigmine to antagonize block produced by rocuronium, vecuronium, atracurium and pancuronium? - Baurain_1996_Br.J.Anaesth_77_496 |
| Author(s) : Baurain MJ , Hoton F , d'Hollander AA , Cantraine FR |
| Ref : British Journal of Anaesthesia , 77 :496 , 1996 |
| Abstract : Baurain_1996_Br.J.Anaesth_77_496 |
| ESTHER : Baurain_1996_Br.J.Anaesth_77_496 |
| PubMedSearch : Baurain_1996_Br.J.Anaesth_77_496 |
| PubMedID: 8942335 |
| Title : Dose-response relationships for neostigmine antagonism of rocuronium-induced neuromuscular block in children and adults - Abdulatif_1996_Br.J.Anaesth_77_710 |
| Author(s) : Abdulatif M , Mowafi H , Al-Ghamdi A , el-Sanabary M |
| Ref : British Journal of Anaesthesia , 77 :710 , 1996 |
| Abstract : Abdulatif_1996_Br.J.Anaesth_77_710 |
| ESTHER : Abdulatif_1996_Br.J.Anaesth_77_710 |
| PubMedSearch : Abdulatif_1996_Br.J.Anaesth_77_710 |
| PubMedID: 9014620 |
| Title : Recovery from mivacurium block with or without anticholinesterase following continuous infusion in obstetric patients - Jan_1996_Anaesth.Intensive.Care_24_585 |
| Author(s) : Jan GS , Tong WN , Chan AM , Hui TW , Lo JW |
| Ref : Anaesthesia & Intensive Care , 24 :585 , 1996 |
| Abstract : Jan_1996_Anaesth.Intensive.Care_24_585 |
| ESTHER : Jan_1996_Anaesth.Intensive.Care_24_585 |
| PubMedSearch : Jan_1996_Anaesth.Intensive.Care_24_585 |
| PubMedID: 8909671 |
| Title : Pseudocholinesterase-mediated hydrolysis is superior to neostigmine for reversal of mivacurium-induced paralysis in vitro - Yang_1996_Anesthesiology_84_936 |
| Author(s) : Yang HS , Goudsouzian N , Martyn JA |
| Ref : Anesthesiology , 84 :936 , 1996 |
| Abstract : Yang_1996_Anesthesiology_84_936 |
| ESTHER : Yang_1996_Anesthesiology_84_936 |
| PubMedSearch : Yang_1996_Anesthesiology_84_936 |
| PubMedID: 8638849 |
| Title : Comparative pharmacokinetics of four cholinesterase inhibitors in rats - Yamamoto_1995_Biol.Pharm.Bull_18_1292 |
| Author(s) : Yamamoto K , Sawada Y , Iga T |
| Ref : Biol Pharm Bull , 18 :1292 , 1995 |
| Abstract : Yamamoto_1995_Biol.Pharm.Bull_18_1292 |
| ESTHER : Yamamoto_1995_Biol.Pharm.Bull_18_1292 |
| PubMedSearch : Yamamoto_1995_Biol.Pharm.Bull_18_1292 |
| PubMedID: 8845827 |
| Title : [Neurotoxic effects of cobra venom (Naja haje haje) on the neuromuscular junction. Electroclinical study of two cases in Tunisia]. [French] - Zouari_1995_Neurophysiologie.Clinique_25_59 |
| Author(s) : Zouari N , Choyakh F |
| Ref : Neurophysiologie Clinique , 25 :59 , 1995 |
| Abstract : Zouari_1995_Neurophysiologie.Clinique_25_59 |
| ESTHER : Zouari_1995_Neurophysiologie.Clinique_25_59 |
| PubMedSearch : Zouari_1995_Neurophysiologie.Clinique_25_59 |
| PubMedID: 7603413 |
| Title : Ventilatory muscle strength and endurance in myasthenia gravis - Keenan_1995_Eur.Respir.J_8_1130 |
| Author(s) : Keenan SP , Alexander D , Road JD , Ryan CF , Oger J , Wilcox PG |
| Ref : European Respiratory Journal , 8 :1130 , 1995 |
| Abstract : Keenan_1995_Eur.Respir.J_8_1130 |
| ESTHER : Keenan_1995_Eur.Respir.J_8_1130 |
| PubMedSearch : Keenan_1995_Eur.Respir.J_8_1130 |
| PubMedID: 7589397 |
| Title : Endogenous acetylcholine-induced Fos expression in magnocellular neurosecretory neurons in the supraoptic nucleus of the rat hypothalamus - Shen_1995_Neurosci.Lett_195_191 |
| Author(s) : Shen E , Sun X |
| Ref : Neuroscience Letters , 195 :191 , 1995 |
| Abstract : Shen_1995_Neurosci.Lett_195_191 |
| ESTHER : Shen_1995_Neurosci.Lett_195_191 |
| PubMedSearch : Shen_1995_Neurosci.Lett_195_191 |
| PubMedID: 8584207 |
| Title : Effect of bethanechol, neostigmine, metoclopramide, and propranolol on myoelectric activity of the ileocecocolic area in cows - Steiner_1995_Am.J.Vet.Res_56_1081 |
| Author(s) : Steiner A , Roussel AJ , Martig J |
| Ref : American Journal of Veterinary Research , 56 :1081 , 1995 |
| Abstract : Steiner_1995_Am.J.Vet.Res_56_1081 |
| ESTHER : Steiner_1995_Am.J.Vet.Res_56_1081 |
| PubMedSearch : Steiner_1995_Am.J.Vet.Res_56_1081 |
| PubMedID: 8533981 |
| Title : Comparison of the effects of neostigmine and edrophonium on the duration of action of suxamethonium - McCoy_1995_Acta.Anaesthesiol.Scand_39_744 |
| Author(s) : McCoy EP , Mirakhur RK |
| Ref : Acta Anaesthesiologica Scandinavica , 39 :744 , 1995 |
| Abstract : McCoy_1995_Acta.Anaesthesiol.Scand_39_744 |
| ESTHER : McCoy_1995_Acta.Anaesthesiol.Scand_39_744 |
| PubMedSearch : McCoy_1995_Acta.Anaesthesiol.Scand_39_744 |
| PubMedID: 7484027 |
| Title : Optimum time for neostigmine administration to antagonize vecuronium-induced neuromuscular blockade - Kirkegaard-Nielsen_1995_Eur.J.Anaesthesiol_12_585 |
| Author(s) : Kirkegaard-Nielsen H , Toft P , Severinsen IK , May O |
| Ref : European Journal of Anaesthesiology , 12 :585 , 1995 |
| Abstract : Kirkegaard-Nielsen_1995_Eur.J.Anaesthesiol_12_585 |
| ESTHER : Kirkegaard-Nielsen_1995_Eur.J.Anaesthesiol_12_585 |
| PubMedSearch : Kirkegaard-Nielsen_1995_Eur.J.Anaesthesiol_12_585 |
| PubMedID: 8665881 |
| Title : The effect of reversal of myoneural blockade on cerebrospinal fluid pressure following cerebral aneurysm surgery - Fawcett_1995_Eur.J.Anaesthesiol_12_591 |
| Author(s) : Fawcett WJ , Chung RA , Fairley CJ , Hollway TE |
| Ref : European Journal of Anaesthesiology , 12 :591 , 1995 |
| Abstract : Fawcett_1995_Eur.J.Anaesthesiol_12_591 |
| ESTHER : Fawcett_1995_Eur.J.Anaesthesiol_12_591 |
| PubMedSearch : Fawcett_1995_Eur.J.Anaesthesiol_12_591 |
| PubMedID: 8665882 |
| Title : Time to peak effect of neostigmine at antagonism of atracurium- or vecuronium-induced neuromuscular block - Kirkegaard-Nielsen_1995_J.Clin.Anesth_7_635 |
| Author(s) : Kirkegaard-Nielsen H , Helbo-Hansen HS , Lindholm P , Severinsen IK , Bulow K |
| Ref : Journal of Clinical Anesthesia , 7 :635 , 1995 |
| Abstract : Kirkegaard-Nielsen_1995_J.Clin.Anesth_7_635 |
| ESTHER : Kirkegaard-Nielsen_1995_J.Clin.Anesth_7_635 |
| PubMedSearch : Kirkegaard-Nielsen_1995_J.Clin.Anesth_7_635 |
| PubMedID: 8747561 |
| Title : Effect of neostigmine at different levels of mivacurium-induced neuromuscular blockade - Trevien_1995_Acta.Anaesth.Scand.Supplementum_106_66 |
| Author(s) : Trevien V , Lienhart A , Just B , Chandon M , Baras E , Camatte S |
| Ref : Acta Anaesthesiologica Scandinavica Supplementum , 106 :66 , 1995 |
| Abstract : Trevien_1995_Acta.Anaesth.Scand.Supplementum_106_66 |
| ESTHER : Trevien_1995_Acta.Anaesth.Scand.Supplementum_106_66 |
| PubMedSearch : Trevien_1995_Acta.Anaesth.Scand.Supplementum_106_66 |
| PubMedID: 8533550 |
| Title : The timing of channel opening during miniature endplate currents at the frog and mouse neuromuscular junctions: effects of fasciculin-2, other anti-cholinesterases and vesamicol - Van der Kloot_1994_Pflugers.Arch_428_114 |
| Author(s) : Van der Kloot W , Balezina OP , Molgo J , Naves LA |
| Ref : Pflugers Arch , 428 :114 , 1994 |
| Abstract : Van der Kloot_1994_Pflugers.Arch_428_114 |
| ESTHER : Van der Kloot_1994_Pflugers.Arch_428_114 |
| PubMedSearch : Van der Kloot_1994_Pflugers.Arch_428_114 |
| PubMedID: 7971167 |
| Title : Degradation of acetylcholine in human airways: role of butyrylcholinesterase - Norel_1993_Br.J.Pharmacol_108_914 |
| Author(s) : Norel X , Angrisani M , Labat C , Gorenne I , Dulmet E , Rossi F , Brink C |
| Ref : British Journal of Pharmacology , 108 :914 , 1993 |
| Abstract : Norel_1993_Br.J.Pharmacol_108_914 |
| ESTHER : Norel_1993_Br.J.Pharmacol_108_914 |
| PubMedSearch : Norel_1993_Br.J.Pharmacol_108_914 |
| PubMedID: 8485630 |
| Title : In vitro protection of acetylcholinesterase and butyrylcholinesterase by tetrahydroaminoacridine. Comparison with physostigmine - Galli_1992_Biochem.Pharmacol_43_2427 |
| Author(s) : Galli A , Mori F , Gori I , Lucherini M |
| Ref : Biochemical Pharmacology , 43 :2427 , 1992 |
| Abstract : Galli_1992_Biochem.Pharmacol_43_2427 |
| ESTHER : Galli_1992_Biochem.Pharmacol_43_2427 |
| PubMedSearch : Galli_1992_Biochem.Pharmacol_43_2427 |
| PubMedID: 1610407 |
| Title : Genetic variants of human serum cholinesterase influence metabolism of the muscle relaxant succinylcholine. - Lockridge_1990_Pharmacol.Ther_47_35 |
| Author(s) : Lockridge O |
| Ref : Pharmacol Ther , 47 :35 , 1990 |
| Abstract : Lockridge_1990_Pharmacol.Ther_47_35 |
| ESTHER : Lockridge_1990_Pharmacol.Ther_47_35 |
| PubMedSearch : Lockridge_1990_Pharmacol.Ther_47_35 |
| PubMedID: 2195556 |
| Gene_locus related to this paper: human-BCHE |
| Title : A peptidase activity exhibited by human serum pseudocholinesterase - Boopathy_1987_Eur.J.Biochem_162_191 |
| Author(s) : Boopathy R , Balasubramanian AS |
| Ref : European Journal of Biochemistry , 162 :191 , 1987 |
| Abstract : Boopathy_1987_Eur.J.Biochem_162_191 |
| ESTHER : Boopathy_1987_Eur.J.Biochem_162_191 |
| PubMedSearch : Boopathy_1987_Eur.J.Biochem_162_191 |
| PubMedID: 3545820 |
| Title : In-vitro and in-vivo protection of acetylcholinesterase by eseroline against inactivation by diisopropyl fluorophosphate and carbamates - Galli_1985_J.Pharm.Pharmacol_37_42 |
| Author(s) : Galli A , Malmberg-Aiello P , Renzi G , Bartolini A |
| Ref : J Pharm Pharmacol , 37 :42 , 1985 |
| Abstract : Galli_1985_J.Pharm.Pharmacol_37_42 |
| ESTHER : Galli_1985_J.Pharm.Pharmacol_37_42 |
| PubMedSearch : Galli_1985_J.Pharm.Pharmacol_37_42 |
| PubMedID: 2858526 |
| Title : Inhibition of human motor endplate cholinesterase by anticholinesterase compounds - Fujii_1982_Acta.Med.Okayama_36_229 |
| Author(s) : Fujii M , Namba T |
| Ref : Acta Med Okayama , 36 :229 , 1982 |
| Abstract : Fujii_1982_Acta.Med.Okayama_36_229 |
| ESTHER : Fujii_1982_Acta.Med.Okayama_36_229 |
| PubMedSearch : Fujii_1982_Acta.Med.Okayama_36_229 |
| PubMedID: 7113748 |
| Title : Activation and blockade of cardiac muscarinic receptors by endogenous acetylcholine and cholinesterase inhibitors - Brown_1982_J.Pharmacol.Exp.Ther_223_20 |
| Author(s) : Brown JH , Wetzel GT , Dunlap J |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 223 :20 , 1982 |
| Abstract : Brown_1982_J.Pharmacol.Exp.Ther_223_20 |
| ESTHER : Brown_1982_J.Pharmacol.Exp.Ther_223_20 |
| PubMedSearch : Brown_1982_J.Pharmacol.Exp.Ther_223_20 |
| PubMedID: 6288918 |
| Title : The aryl acylamidases and their relationship to cholinesterases in human serum, erythrocyte and liver - George_1981_Eur.J.Biochem_121_177 |
| Author(s) : George ST , Balasubramanian AS |
| Ref : European Journal of Biochemistry , 121 :177 , 1981 |
| Abstract : George_1981_Eur.J.Biochem_121_177 |
| ESTHER : George_1981_Eur.J.Biochem_121_177 |
| PubMedSearch : George_1981_Eur.J.Biochem_121_177 |
| PubMedID: 7035166 |
| Title : Differential inhibition of plasma cholinesterase variants using the dibutyrate analogue of pancuronium bromide - Whittaker_1981_Hum.Hered_31_242 |
| Author(s) : Whittaker M , Britten JJ |
| Ref : Hum Hered , 31 :242 , 1981 |
| Abstract : Whittaker_1981_Hum.Hered_31_242 |
| ESTHER : Whittaker_1981_Hum.Hered_31_242 |
| PubMedSearch : Whittaker_1981_Hum.Hered_31_242 |
| PubMedID: 7287015 |
| Title : The identity of the serotonin-sensitive aryl acylamidase with acetylcholinesterase from human erythrocytes, sheep basal ganglia and electric eel - George_1980_Eur.J.Biochem_111_511 |
| Author(s) : George ST , Balasubramanian AS |
| Ref : European Journal of Biochemistry , 111 :511 , 1980 |
| Abstract : George_1980_Eur.J.Biochem_111_511 |
| ESTHER : George_1980_Eur.J.Biochem_111_511 |
| PubMedSearch : George_1980_Eur.J.Biochem_111_511 |
| PubMedID: 7460914 |
| Title : The inhibition of frog tissue cholinesterases and the influence of eserine and prostigmine on the action of acetylcholine on the frog heart - Majcen_1975_Biochem.Pharmacol_24_1599 |
| Author(s) : Majcen Z , Brzin M |
| Ref : Biochemical Pharmacology , 24 :1599 , 1975 |
| Abstract : Majcen_1975_Biochem.Pharmacol_24_1599 |
| ESTHER : Majcen_1975_Biochem.Pharmacol_24_1599 |
| PubMedSearch : Majcen_1975_Biochem.Pharmacol_24_1599 |
| PubMedID: 1081394 |