Ar9281
inhibitor developed by Arete Therapeutics as investigational new drug (IND) candidate that has reached Phase I clinical trial for hypertension: was unable to demonstrate efficacy in human patients.
General
Type : Adamantyl,Urea derivative
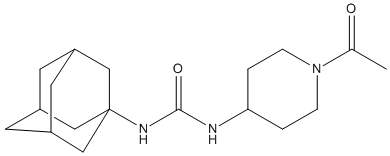
Chemical_Nomenclature : 1-(1-acetylpiperidin-4-yl)-3-(1-adamantyl)urea
Canonical SMILES : CC(=O)N1CCC(CC1)NC(=O)NC23CC4CC(C2)CC(C4)C3
InChI : InChI=1S\/C18H29N3O2\/c1-12(22)21-4-2-16(3-5-21)19-17(23)20-18-9-13-6-14(10-18)8-15(7-13)11-18\/h13-16H,2-11H2,1H3,(H2,19,20,23)
InChIKey : HUDQLWBKJOMXSZ-UHFFFAOYSA-N
Other name(s) : UNII-4HA03Q8EZ9,Ar-9281,4HA03Q8EZ9,CHEMBL436774,1-(Adamantane-1-yl)-3-(1-acetylpiperidine-4-yl)urea,1-[(1-Acetylpiperidin-4-yl)-3-adamantan-1-yl]urea
MW : 319.44
Formula : C18H29N3O2
CAS_number : 913548-29-5
PubChem : 12000797
UniChem : HUDQLWBKJOMXSZ-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
References (7)
| Title : Identification and optimization of soluble epoxide hydrolase inhibitors with dual potency towards fatty acid amide hydrolase - Kodani_2018_Bioorg.Med.Chem.Lett_28_762 |
| Author(s) : Kodani SD , Bhakta S , Hwang SH , Pakhomova S , Newcomer ME , Morisseau C , Hammock BD |
| Ref : Bioorganic & Medicinal Chemistry Lett , 28 :762 , 2018 |
| Abstract : Kodani_2018_Bioorg.Med.Chem.Lett_28_762 |
| ESTHER : Kodani_2018_Bioorg.Med.Chem.Lett_28_762 |
| PubMedSearch : Kodani_2018_Bioorg.Med.Chem.Lett_28_762 |
| PubMedID: 29366648 |
| Gene_locus related to this paper: human-EPHX2 |
| Title : Pharmacological inhibition of soluble epoxide hydrolase provides cardioprotection in hyperglycemic rats - Guglielmino_2012_Am.J.Physiol.Heart.Circ.Physiol_303_H853 |
| Author(s) : Guglielmino K , Jackson K , Harris TR , Vu V , Dong H , Dutrow G , Evans JE , Graham J , Cummings BP , Havel PJ , Chiamvimonvat N , Despa S , Hammock BD , Despa F |
| Ref : American Journal of Physiology Heart Circ Physiol , 303 :H853 , 2012 |
| Abstract : Guglielmino_2012_Am.J.Physiol.Heart.Circ.Physiol_303_H853 |
| ESTHER : Guglielmino_2012_Am.J.Physiol.Heart.Circ.Physiol_303_H853 |
| PubMedSearch : Guglielmino_2012_Am.J.Physiol.Heart.Circ.Physiol_303_H853 |
| PubMedID: 22865388 |
| Title : Inhibition of soluble epoxide hydrolase reduces food intake and increases metabolic rate in obese mice - do Carmo_2012_Nutr.Metab.Cardiovasc.Dis_22_598 |
| Author(s) : do Carmo JM , da Silva AA , Morgan J , Jim Wang YX , Munusamy S , Hall JE |
| Ref : Nutr Metab Cardiovasc Dis , 22 :598 , 2012 |
| Abstract : do Carmo_2012_Nutr.Metab.Cardiovasc.Dis_22_598 |
| ESTHER : do Carmo_2012_Nutr.Metab.Cardiovasc.Dis_22_598 |
| PubMedSearch : do Carmo_2012_Nutr.Metab.Cardiovasc.Dis_22_598 |
| PubMedID: 21190818 |
| Title : Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects - Chen_2012_J.Clin.Pharmacol_52_319 |
| Author(s) : Chen D , Whitcomb R , MacIntyre E , Tran V , Do ZN , Sabry J , Patel DV , Anandan SK , Gless R , Webb HK |
| Ref : Journal of Clinical Pharmacology , 52 :319 , 2012 |
| Abstract : Chen_2012_J.Clin.Pharmacol_52_319 |
| ESTHER : Chen_2012_J.Clin.Pharmacol_52_319 |
| PubMedSearch : Chen_2012_J.Clin.Pharmacol_52_319 |
| PubMedID: 21422238 |
| Title : Soluble epoxide hydrolase inhibitors and their potential for treatment of multiple pathologic conditions - Ingraham_2011_Curr.Med.Chem_18_587 |
| Author(s) : Ingraham RH , Gless RD , Lo HY |
| Ref : Curr Med Chem , 18 :587 , 2011 |
| Abstract : Ingraham_2011_Curr.Med.Chem_18_587 |
| ESTHER : Ingraham_2011_Curr.Med.Chem_18_587 |
| PubMedSearch : Ingraham_2011_Curr.Med.Chem_18_587 |
| PubMedID: 21143109 |
| Title : Inhibition of soluble epoxide hydrolase attenuates endothelial dysfunction in animal models of diabetes, obesity and hypertension - Zhang_2011_Eur.J.Pharmacol_654_68 |
| Author(s) : Zhang LN , Vincelette J , Chen D , Gless RD , Anandan SK , Rubanyi GM , Webb HK , MacIntyre DE , Wang YX |
| Ref : European Journal of Pharmacology , 654 :68 , 2011 |
| Abstract : Zhang_2011_Eur.J.Pharmacol_654_68 |
| ESTHER : Zhang_2011_Eur.J.Pharmacol_654_68 |
| PubMedSearch : Zhang_2011_Eur.J.Pharmacol_654_68 |
| PubMedID: 21187082 |
| Title : 1-(1-acetyl-piperidin-4-yl)-3-adamantan-1-yl-urea (AR9281) as a potent, selective, and orally available soluble epoxide hydrolase inhibitor with efficacy in rodent models of hypertension and dysglycemia - Anandan_2011_Bioorg.Med.Chem.Lett_21_983 |
| Author(s) : Anandan SK , Webb HK , Chen D , Wang YX , Aavula BR , Cases S , Cheng Y , Do ZN , Mehra U , Tran V , Vincelette J , Waszczuk J , White K , Wong KR , Zhang LN , Jones PD , Hammock BD , Patel DV , Whitcomb R , MacIntyre DE , Sabry J , Gless R |
| Ref : Bioorganic & Medicinal Chemistry Lett , 21 :983 , 2011 |
| Abstract : Anandan_2011_Bioorg.Med.Chem.Lett_21_983 |
| ESTHER : Anandan_2011_Bioorg.Med.Chem.Lett_21_983 |
| PubMedSearch : Anandan_2011_Bioorg.Med.Chem.Lett_21_983 |
| PubMedID: 21211973 |