Gallamine~Flaxedil
M2 muscarinic receptor antagonist; muscle relaxant . Derivative of pyrogallol pyrogallic acid || Skeletal muscle relaxant
General
Type : Neuromuscular Nondepolarizing Agents,Nicotinic antagonist,Not A\/B H target
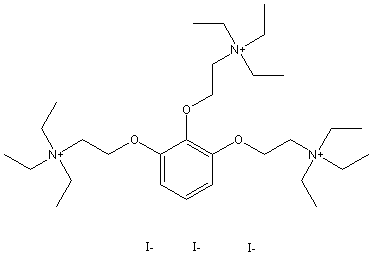
Chemical_Nomenclature : 2-[2,3-bis[2-(triethylazaniumyl)ethoxy]phenoxy]ethyl-triethylazanium\;triiodide
Canonical SMILES : CC[N+](CC)(CC)CCOC1=C(C(=CC=C1)OCC[N+](CC)(CC)CC)OCC[N+](CC)(CC)CC.[I-].[I-].[I-]
InChI : InChI=1S\/C30H60N3O3.3HI\/c1-10-31(11-2,12-3)22-25-34-28-20-19-21-29(35-26-23-32(13-4,14-5)15-6)30(28)36-27-24-33(16-7,17-8)18-9\;\;\;\/h19-21H,10-18,22-27H2,1-9H3\;3*1H\/q+3\;\;\;\/p-3
InChIKey : REEUVFCVXKWOFE-UHFFFAOYSA-K
Other name(s) : 2,2',2'-[1,2,3 Benzene-triyltris(oxy)]tris[N,N,N-triethylethanaminium] triiodide,[v-Phenenyltris(oxyethylene)]tris[triethylammonium iodide],1,2,3- Tris(2-triethylammonium ethoxy)benzene triiodide,1,2,3-Tris(2-diethylaminoethoxy)benzene triethiodide,1,2,3-Tris(2-diethylaminoethoxy)benzene tris(ethyliodide),Tri(diethylaminoethoxy)-1,2,3-benzene triiodoethylate,Tricuran,1,2,3-Tri(beta- diethylaminoethoxy)benzene triethiodide,1,2,3-Tris(diethylaminoethoxy)benzene triethiodide,Pyrogallol 1,2,3-tris(diethylaminoethyl) ether tris(ethyliodide),Tri(beta-diethylaminoethoxy)-1,2,3-benzene triiodoethylate,GMN,Gallamine triethiodide,Retensin,Relaxan,Flaxedil,Sincurarine,Syncurarine,Benzcurine,Benzkurin,Flacedil,Gallaflex,Gallamine,triiodoethylate,Gallamine-3EtI,Miowas G,Parexyl,Pirolakson,Pyrolaxon,Remyolan
MW : 891.56
Formula : C30H60I3N3O3
CAS_number : 65-29-2
PubChem : 6172
UniChem : REEUVFCVXKWOFE-UHFFFAOYSA-K
IUPHAR : 356
Wikipedia : Gallamine_triethiodide
Target
Families : Gallamine~Flaxedil ligand of proteins in family: ACHE || BCHE
Stucture : 1N5M Crystal structure of the mouse acetylcholinesterase-gallamine complex
Protein : mouse-ACHE
References (10)
| Title : The influence of peripheral site ligands on the reaction of symmetric and chiral organophosphates with wildtype and mutant acetylcholinesterases - Radic_1999_Chem.Biol.Interact_119-120_111 |
| Author(s) : Radic Z , Taylor P |
| Ref : Chemico-Biological Interactions , 119-120 :111 , 1999 |
| Abstract : Radic_1999_Chem.Biol.Interact_119-120_111 |
| ESTHER : Radic_1999_Chem.Biol.Interact_119-120_111 |
| PubMedSearch : Radic_1999_Chem.Biol.Interact_119-120_111 |
| PubMedID: 10421444 |
| Title : Nonequilibrium analysis alters the mechanistic interpretation of inhibition of acetylcholinesterase by peripheral site ligands - Szegletes_1998_Biochemistry_37_4206 |
| Author(s) : Szegletes T , Mallender WD , Rosenberry TL |
| Ref : Biochemistry , 37 :4206 , 1998 |
| Abstract : Szegletes_1998_Biochemistry_37_4206 |
| ESTHER : Szegletes_1998_Biochemistry_37_4206 |
| PubMedSearch : Szegletes_1998_Biochemistry_37_4206 |
| PubMedID: 9521743 |
| Title : The inhibitory effect of the neuromuscular blocking agent, gallamine triethiodide, on camel retina acetylcholinesterase activity - Al-Jafari_1997_Toxicol.Lett_90_45 |
| Author(s) : Al-Jafari AA |
| Ref : Toxicology Letters , 90 :45 , 1997 |
| Abstract : Al-Jafari_1997_Toxicol.Lett_90_45 |
| ESTHER : Al-Jafari_1997_Toxicol.Lett_90_45 |
| PubMedSearch : Al-Jafari_1997_Toxicol.Lett_90_45 |
| PubMedID: 9020401 |
| Title : Cloning and expression of acetylcholinesterase from Bungarus fasciatus venom. A new type of cooh-terminal domain\; involvement of a positively charged residue in the peripheral site - Cousin_1996_J.Biol.Chem_271_15099 |
| Author(s) : Cousin X , Bon S , Duval N , Massoulie J , Bon C |
| Ref : Journal of Biological Chemistry , 271 :15099 , 1996 |
| Abstract : Cousin_1996_J.Biol.Chem_271_15099 |
| ESTHER : Cousin_1996_J.Biol.Chem_271_15099 |
| PubMedSearch : Cousin_1996_J.Biol.Chem_271_15099 |
| PubMedID: 8662867 |
| Gene_locus related to this paper: bunfa-ACHE |
| Title : Allosteric regulation of the binding of [3H]acetylcholine to m2 muscarinic receptors - Gnagey_1996_Biochem.Pharmacol_52_1767 |
| Author(s) : Gnagey AL , Ellis J |
| Ref : Biochemical Pharmacology , 52 :1767 , 1996 |
| Abstract : Gnagey_1996_Biochem.Pharmacol_52_1767 |
| ESTHER : Gnagey_1996_Biochem.Pharmacol_52_1767 |
| PubMedSearch : Gnagey_1996_Biochem.Pharmacol_52_1767 |
| PubMedID: 8986140 |
| Title : Differential effects of peripheral site ligands on Torpedo and chicken acetylcholinesterase - Eichler_1994_Mol.Pharmacol_45_335 |
| Author(s) : Eichler J , Anselmet A , Sussman JL , Massoulie J , Silman I |
| Ref : Molecular Pharmacology , 45 :335 , 1994 |
| Abstract : Eichler_1994_Mol.Pharmacol_45_335 |
| ESTHER : Eichler_1994_Mol.Pharmacol_45_335 |
| PubMedSearch : Eichler_1994_Mol.Pharmacol_45_335 |
| PubMedID: 8114681 |
| Title : Non-reactivating effects of HI-6 on hippocampal neurotransmission - Melchers_1994_Arch.Toxicol_69_118 |
| Author(s) : Melchers BP , van der Laaken AL , Busker RW , Bruijnzeel PL , van Helden HP |
| Ref : Archives of Toxicology , 69 :118 , 1994 |
| Abstract : Melchers_1994_Arch.Toxicol_69_118 |
| ESTHER : Melchers_1994_Arch.Toxicol_69_118 |
| PubMedSearch : Melchers_1994_Arch.Toxicol_69_118 |
| PubMedID: 7717860 |
| Title : Acetylcholinesterase as polyelectrolyte: interaction with multivalent cationic inhibitors - Tougu_1993_Biochim.Biophys.Acta_1157_199 |
| Author(s) : Tougu V , Kesvatera T , Laane A , Aaviksaar A |
| Ref : Biochimica & Biophysica Acta , 1157 :199 , 1993 |
| Abstract : Tougu_1993_Biochim.Biophys.Acta_1157_199 |
| ESTHER : Tougu_1993_Biochim.Biophys.Acta_1157_199 |
| PubMedSearch : Tougu_1993_Biochim.Biophys.Acta_1157_199 |
| PubMedID: 8099499 |
| Title : Structure-activity relationship of reversible cholinesterase inhibitors including paraquat - Seto_1988_Arch.Toxicol_62_37 |
| Author(s) : Seto Y , Shinohara T |
| Ref : Archives of Toxicology , 62 :37 , 1988 |
| Abstract : Seto_1988_Arch.Toxicol_62_37 |
| ESTHER : Seto_1988_Arch.Toxicol_62_37 |
| PubMedSearch : Seto_1988_Arch.Toxicol_62_37 |
| PubMedID: 3190453 |
| Title : Interaction of fluorescence probes with acetylcholinesterase. The site and specificity of propidium binding - Taylor_1975_Biochemistry_14_1989 |
| Author(s) : Taylor P , Lappi S |
| Ref : Biochemistry , 14 :1989 , 1975 |
| Abstract : Taylor_1975_Biochemistry_14_1989 |
| ESTHER : Taylor_1975_Biochemistry_14_1989 |
| PubMedSearch : Taylor_1975_Biochemistry_14_1989 |
| PubMedID: 1125207 |