Hexamethonium
A nicotinic cholinergic antagonist often referred to as the prototypical ganglionic blocker. It is poorly absorbed from the gastrointestinal tract and does not cross the blood-brain barrier. It has been used for a variety of therapeutic purposes including hypertension but, like the other ganglionic blockers, it has been replaced by more specific drugs for most purposes, although it is widely used a research tool
General
Type : Bisquaternary,Nicotinic antagonist,Trimethylammonium
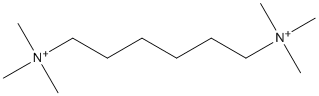
Chemical_Nomenclature : 1,6-bis(trimethylammonium)hexane
Canonical SMILES : C[N+](C)(C)CCCCCC[N+](C)(C)C.[OH-].[OH-]
InChI : InChI=1S\/C12H30N2.2H2O\/c1-13(2,3)11-9-7-8-10-12-14(4,5)6\;\;\/h7-12H2,1-6H3\;2*1H2\/q+2\;\;\/p-2
InChIKey : GYLUMIIRFKDCKI-UHFFFAOYSA-L
Other name(s) : UNII-RVL393940Z,Hexamethonium hydroxide solution,Hexamethonum,Bistrium,Depressin,Hexanium
MW : 273.29
Formula : C12H30N2Cl2
CAS_number : 60-25-3
PubChem : 6059
UniChem : GYLUMIIRFKDCKI-UHFFFAOYSA-L
IUPHAR :
Wikipedia : Hexamethonium
Target
References (30)
| Title : Subchronic effects following a single sarin exposure on blood-brain and blood-testes barrier permeability, acetylcholinesterase, and acetylcholine receptors in the central nervous system of rat: a dose-response study - Jones_2000_J.Toxicol.Environ.Health.A_61_695 |
| Author(s) : Jones KH , Dechkovskaia AM , Herrick EA , Abdel-Rahman AA , Khan WA , Abou-Donia MB |
| Ref : J Toxicol Environ Health A , 61 :695 , 2000 |
| Abstract : Jones_2000_J.Toxicol.Environ.Health.A_61_695 |
| ESTHER : Jones_2000_J.Toxicol.Environ.Health.A_61_695 |
| PubMedSearch : Jones_2000_J.Toxicol.Environ.Health.A_61_695 |
| PubMedID: 11132698 |
| Title : Indian red scorpion venom modulates spontaneous activity of rat right atria through the involvement of cholinergic and adrenergic systems - Alex_1999_Indian.J.Exp.Biol_37_455 |
| Author(s) : Alex AB , Deshpande SB |
| Ref : Indian J Exp Biol , 37 :455 , 1999 |
| Abstract : Alex_1999_Indian.J.Exp.Biol_37_455 |
| ESTHER : Alex_1999_Indian.J.Exp.Biol_37_455 |
| PubMedSearch : Alex_1999_Indian.J.Exp.Biol_37_455 |
| PubMedID: 10492617 |
| Title : Increased plasma cholinesterase activity and mivacurium resistance: report of a family - |
| Author(s) : Naguib M , Gomaa M , Samarkandi AH , Bevan DR , Akkielah AK , Watson C , Billecke S , La Du BN |
| Ref : Anesthesia & Analgesia , 89 :1579 , 1999 |
| PubMedID: 10589654 |
| Title : Cholinergic nerve function in monkey ciliary arteries innervated by nitroxidergic nerve - Toda_1998_Am.J.Physiol_274_H1582 |
| Author(s) : Toda N , Toda M , Ayajiki K , Okamura T |
| Ref : American Journal of Physiology , 274 :H1582 , 1998 |
| Abstract : Toda_1998_Am.J.Physiol_274_H1582 |
| ESTHER : Toda_1998_Am.J.Physiol_274_H1582 |
| PubMedSearch : Toda_1998_Am.J.Physiol_274_H1582 |
| PubMedID: 9612367 |
| Title : Effects of hexamethonium, phenothiazines, propranolol and ephedrine on acetylcholinesterase carbamylation by physostigmine, aldicarb and carbaryl: interaction between the active site and the functionally distinct peripheral sites in acetylcholinesterase - Singh_1998_Comp.Biochem.Physiol.C.Pharmacol.Toxicol.Endocrinol_119_97 |
| Author(s) : Singh AK , Spassova D |
| Ref : Comparative Biochemistry & Physiology C Pharmacology Toxicology & Endocrinology , 119 :97 , 1998 |
| Abstract : Singh_1998_Comp.Biochem.Physiol.C.Pharmacol.Toxicol.Endocrinol_119_97 |
| ESTHER : Singh_1998_Comp.Biochem.Physiol.C.Pharmacol.Toxicol.Endocrinol_119_97 |
| PubMedSearch : Singh_1998_Comp.Biochem.Physiol.C.Pharmacol.Toxicol.Endocrinol_119_97 |
| PubMedID: 9568379 |
| Title : Pharmacology of neurotransmission to the smooth muscle of the rat and the guinea-pig prostate glands - Lau_1998_J.Auton.Pharmacol_18_349 |
| Author(s) : Lau WA , Ventura S , Pennefather JN |
| Ref : J Auton Pharmacol , 18 :349 , 1998 |
| Abstract : Lau_1998_J.Auton.Pharmacol_18_349 |
| ESTHER : Lau_1998_J.Auton.Pharmacol_18_349 |
| PubMedSearch : Lau_1998_J.Auton.Pharmacol_18_349 |
| PubMedID: 9915599 |
| Title : Basal acetylcholine release in leech ganglia depolarizes neurons through receptors with a nicotinic binding site - Marin Burgin_1998_J.Exp.Biol_201_1907 |
| Author(s) : Marin Burgin A , Szczupak L |
| Ref : J Exp Biol , 201 :1907 , 1998 |
| Abstract : Marin Burgin_1998_J.Exp.Biol_201_1907 |
| ESTHER : Marin Burgin_1998_J.Exp.Biol_201_1907 |
| PubMedSearch : Marin Burgin_1998_J.Exp.Biol_201_1907 |
| PubMedID: 9722429 |
| Title : Bradycardia produced by pyridostigmine and physostigmine - Stein_1997_Can.J.Anaesth_44_1286 |
| Author(s) : Stein RD , Backman SB , Collier B , Polosa C |
| Ref : Canadian Journal of Anaesthesia , 44 :1286 , 1997 |
| Abstract : Stein_1997_Can.J.Anaesth_44_1286 |
| ESTHER : Stein_1997_Can.J.Anaesth_44_1286 |
| PubMedSearch : Stein_1997_Can.J.Anaesth_44_1286 |
| PubMedID: 9429048 |
| Title : Evaluation of the nature of camel retinal acetylcholinesterase: inhibition by hexamethonium - Alhomida_1997_J.Enzyme.Inhib_12_303 |
| Author(s) : Alhomida AS , Kamal MA , Al-Jafari AA |
| Ref : J Enzyme Inhib , 12 :303 , 1997 |
| Abstract : Alhomida_1997_J.Enzyme.Inhib_12_303 |
| ESTHER : Alhomida_1997_J.Enzyme.Inhib_12_303 |
| PubMedSearch : Alhomida_1997_J.Enzyme.Inhib_12_303 |
| PubMedID: 9502051 |
| Title : Is the input to a GABAergic or cholinergic synapse the sole asymmetry in rabbit's retinal directional selectivity? - Grzymacz_1997_Visual.Neurosci_14_39 |
| Author(s) : Grzymacz NM , Tootle JS , Amthor FR |
| Ref : Visual Neuroscience , 14 :39 , 1997 |
| Abstract : Grzymacz_1997_Visual.Neurosci_14_39 |
| ESTHER : Grzymacz_1997_Visual.Neurosci_14_39 |
| PubMedSearch : Grzymacz_1997_Visual.Neurosci_14_39 |
| PubMedID: 9057267 |
| Title : Possible nicotinic receptor-mediated modulation of synaptic transmission in nucleus of the solitary tract - Shiraki_1997_Am.J.Physiol_272_R869 |
| Author(s) : Shiraki T , Toyoda A , Sugino H , Hori A , Kobayashi S |
| Ref : American Journal of Physiology , 272 :R869 , 1997 |
| Abstract : Shiraki_1997_Am.J.Physiol_272_R869 |
| ESTHER : Shiraki_1997_Am.J.Physiol_272_R869 |
| PubMedSearch : Shiraki_1997_Am.J.Physiol_272_R869 |
| PubMedID: 9087649 |
| Title : Cardiovascular effects of an organophosphate toxin isolated from Ptychodiscus brevis - Mazumder_1997_Biomed.Environ.Sci_10_85 |
| Author(s) : Mazumder PK , Gupta AK , Kaushik MP , Kumar D , Dube SN |
| Ref : Biomedical & Environmental Sciences , 10 :85 , 1997 |
| Abstract : Mazumder_1997_Biomed.Environ.Sci_10_85 |
| ESTHER : Mazumder_1997_Biomed.Environ.Sci_10_85 |
| PubMedSearch : Mazumder_1997_Biomed.Environ.Sci_10_85 |
| PubMedID: 9099430 |
| Title : Central nicotinic receptor blockade inhibits emotionally conditioned pressor responses in rats - Kubo_1996_Experientia_52_348 |
| Author(s) : Kubo T , Katsumata Y , Fukumori R , Taguchi K , Hagiwara Y |
| Ref : Experientia , 52 :348 , 1996 |
| Abstract : Kubo_1996_Experientia_52_348 |
| ESTHER : Kubo_1996_Experientia_52_348 |
| PubMedSearch : Kubo_1996_Experientia_52_348 |
| PubMedID: 8620939 |
| Title : [Effects of drugs on re-excitability of synapses of isolated frog ganglia] - Sandorne_1996_Acta.Pharm.Hung_66_259 |
| Author(s) : Sandorne KM , Blazso G |
| Ref : Acta Pharm Hung , 66 :259 , 1996 |
| Abstract : Sandorne_1996_Acta.Pharm.Hung_66_259 |
| ESTHER : Sandorne_1996_Acta.Pharm.Hung_66_259 |
| PubMedSearch : Sandorne_1996_Acta.Pharm.Hung_66_259 |
| PubMedID: 9604492 |
| Title : Acute toxicity of several organophosphorous insecticides and protection by cholinergic antagonists and 2-PAM on Artemia salina larvae - Sanchez-Fortun_1996_Arch.Environ.Contam.Toxicol_31_391 |
| Author(s) : Sanchez-Fortun S , Sanz F , Barahona MV |
| Ref : Archives of Environmental Contamination & Toxicology , 31 :391 , 1996 |
| Abstract : Sanchez-Fortun_1996_Arch.Environ.Contam.Toxicol_31_391 |
| ESTHER : Sanchez-Fortun_1996_Arch.Environ.Contam.Toxicol_31_391 |
| PubMedSearch : Sanchez-Fortun_1996_Arch.Environ.Contam.Toxicol_31_391 |
| PubMedID: 8854833 |
| Title : Use of C1300 neuroblastoma cells to evaluate the protective value of hexamethonium, trimethaphan, hemicholinium, and triethylcholine against diisopropyl phosphorofluoridate toxicity - Hong_1995_J.Pharmaceutical.Scis_84_65 |
| Author(s) : Hong SJ , Rohde BH , Chiou GC |
| Ref : Journal of Pharmaceutical Sciences , 84 :65 , 1995 |
| Abstract : Hong_1995_J.Pharmaceutical.Scis_84_65 |
| ESTHER : Hong_1995_J.Pharmaceutical.Scis_84_65 |
| PubMedSearch : Hong_1995_J.Pharmaceutical.Scis_84_65 |
| PubMedID: 7714747 |
| Title : Differential effects of peripheral site ligands on Torpedo and chicken acetylcholinesterase - Eichler_1994_Mol.Pharmacol_45_335 |
| Author(s) : Eichler J , Anselmet A , Sussman JL , Massoulie J , Silman I |
| Ref : Molecular Pharmacology , 45 :335 , 1994 |
| Abstract : Eichler_1994_Mol.Pharmacol_45_335 |
| ESTHER : Eichler_1994_Mol.Pharmacol_45_335 |
| PubMedSearch : Eichler_1994_Mol.Pharmacol_45_335 |
| PubMedID: 8114681 |
| Title : Histochemical and functional evidence for a cholinergic innervation of the equine ureter - Prieto_1994_J.Autonom.Nerv.Syst_47_159 |
| Author(s) : Prieto D , Simonsen U , Martin J , Hernandez M , Rivera L , Lema L , Garcia P , Garcia-Sacristan A |
| Ref : Journal of the Autonomic Nervous System , 47 :159 , 1994 |
| Abstract : Prieto_1994_J.Autonom.Nerv.Syst_47_159 |
| ESTHER : Prieto_1994_J.Autonom.Nerv.Syst_47_159 |
| PubMedSearch : Prieto_1994_J.Autonom.Nerv.Syst_47_159 |
| PubMedID: 7912246 |
| Title : Acetylcholinesterase peripheral anionic site degeneracy conferred by amino acid arrays sharing a common core - Barak_1994_J.Biol.Chem_269_6296 |
| Author(s) : Barak D , Kronman C , Ordentlich A , Ariel N , Bromberg A , Marcus D , Lazar A , Velan B , Shafferman A |
| Ref : Journal of Biological Chemistry , 269 :6296 , 1994 |
| Abstract : Barak_1994_J.Biol.Chem_269_6296 |
| ESTHER : Barak_1994_J.Biol.Chem_269_6296 |
| PubMedSearch : Barak_1994_J.Biol.Chem_269_6296 |
| PubMedID: 8119978 |
| Title : Acetylcholinesterase as polyelectrolyte: interaction with multivalent cationic inhibitors - Tougu_1993_Biochim.Biophys.Acta_1157_199 |
| Author(s) : Tougu V , Kesvatera T , Laane A , Aaviksaar A |
| Ref : Biochimica & Biophysica Acta , 1157 :199 , 1993 |
| Abstract : Tougu_1993_Biochim.Biophys.Acta_1157_199 |
| ESTHER : Tougu_1993_Biochim.Biophys.Acta_1157_199 |
| PubMedSearch : Tougu_1993_Biochim.Biophys.Acta_1157_199 |
| PubMedID: 8099499 |
| Title : Inhibition of acetyl- and butyrylcholinesterase and amylase release from canine pancreas - Oguchi_1989_Pancreas_4_423 |
| Author(s) : Oguchi Y , Dressel TD , Borner JW , Miller J , Goodale RL |
| Ref : Pancreas , 4 :423 , 1989 |
| Abstract : Oguchi_1989_Pancreas_4_423 |
| ESTHER : Oguchi_1989_Pancreas_4_423 |
| PubMedSearch : Oguchi_1989_Pancreas_4_423 |
| PubMedID: 2474812 |
| Title : Secretion of acetylcholinesterase and butyrylcholinesterase from the guinea-pig isolated ileum - Appleyard_1989_Br.J.Pharmacol_97_490 |
| Author(s) : Appleyard ME , Smith AD |
| Ref : British Journal of Pharmacology , 97 :490 , 1989 |
| Abstract : Appleyard_1989_Br.J.Pharmacol_97_490 |
| ESTHER : Appleyard_1989_Br.J.Pharmacol_97_490 |
| PubMedSearch : Appleyard_1989_Br.J.Pharmacol_97_490 |
| PubMedID: 2758227 |
| Title : Effects of some mono- and bisquaternary ammonium compounds on the reactivatability of soman-inhibited human acetylcholinesterase in vitro - Hallek_1988_Biochem.Pharmacol_37_819 |
| Author(s) : Hallek M , Szinicz L |
| Ref : Biochemical Pharmacology , 37 :819 , 1988 |
| Abstract : Hallek_1988_Biochem.Pharmacol_37_819 |
| ESTHER : Hallek_1988_Biochem.Pharmacol_37_819 |
| PubMedSearch : Hallek_1988_Biochem.Pharmacol_37_819 |
| PubMedID: 3345199 |
| Title : Structure-activity relationship of reversible cholinesterase inhibitors including paraquat - Seto_1988_Arch.Toxicol_62_37 |
| Author(s) : Seto Y , Shinohara T |
| Ref : Archives of Toxicology , 62 :37 , 1988 |
| Abstract : Seto_1988_Arch.Toxicol_62_37 |
| ESTHER : Seto_1988_Arch.Toxicol_62_37 |
| PubMedSearch : Seto_1988_Arch.Toxicol_62_37 |
| PubMedID: 3190453 |
| Title : Effects of cholinergic drugs on muscle contraction in Moniliformis moniliformis (Acanthocephala) - Sangster_1987_J.Parasitol_73_998 |
| Author(s) : Sangster NC , Mettrick DF |
| Ref : J Parasitol , 73 :998 , 1987 |
| Abstract : Sangster_1987_J.Parasitol_73_998 |
| ESTHER : Sangster_1987_J.Parasitol_73_998 |
| PubMedSearch : Sangster_1987_J.Parasitol_73_998 |
| PubMedID: 3656016 |
| Title : Selective facilitatory effect of vasoactive intestinal polypeptide (VIP) on muscarinic firing in vesical ganglia of the cat - Kawatani_1985_Brain.Res_336_223 |
| Author(s) : Kawatani M , Rutigliano M , De Groat WC |
| Ref : Brain Research , 336 :223 , 1985 |
| Abstract : Kawatani_1985_Brain.Res_336_223 |
| ESTHER : Kawatani_1985_Brain.Res_336_223 |
| PubMedSearch : Kawatani_1985_Brain.Res_336_223 |
| PubMedID: 3891019 |
| Title : Efficacy and toxicity of drug combinations in treatment of physostigmine toxicosis - Klemm_1983_Toxicology_27_41 |
| Author(s) : Klemm WR |
| Ref : Toxicology , 27 :41 , 1983 |
| Abstract : Klemm_1983_Toxicology_27_41 |
| ESTHER : Klemm_1983_Toxicology_27_41 |
| PubMedSearch : Klemm_1983_Toxicology_27_41 |
| PubMedID: 6149635 |
| Title : Effects of organophosphate insecticides on the cholinergic receptors of frog skeletal muscle - Dekin_1978_J.Pharmacol.Exp.Ther_205_319 |
| Author(s) : Dekin MS , Guy HR , Edwards C |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 205 :319 , 1978 |
| Abstract : Dekin_1978_J.Pharmacol.Exp.Ther_205_319 |
| ESTHER : Dekin_1978_J.Pharmacol.Exp.Ther_205_319 |
| PubMedSearch : Dekin_1978_J.Pharmacol.Exp.Ther_205_319 |
| PubMedID: 641831 |
| Title : The ganglionic blocking properties of the cholinesterase reactivator, HS-6 - Lundy_1978_Can.J.Physiol.Pharmacol_56_857 |
| Author(s) : Lundy PM |
| Ref : Canadian Journal of Physiology & Pharmacology , 56 :857 , 1978 |
| Abstract : Lundy_1978_Can.J.Physiol.Pharmacol_56_857 |
| ESTHER : Lundy_1978_Can.J.Physiol.Pharmacol_56_857 |
| PubMedSearch : Lundy_1978_Can.J.Physiol.Pharmacol_56_857 |
| PubMedID: 30527 |
| Title : Differences in subunit activities in acetylcholinesterase as possible cause for apparent deviation from normal Michaelis-Menten kinetics - Gentinetta_1976_Biochim.Biophys.Acta_438_437 |
| Author(s) : Gentinetta R , Brodbeck U |
| Ref : Biochimica & Biophysica Acta , 438 :437 , 1976 |
| Abstract : Gentinetta_1976_Biochim.Biophys.Acta_438_437 |
| ESTHER : Gentinetta_1976_Biochim.Biophys.Acta_438_437 |
| PubMedSearch : Gentinetta_1976_Biochim.Biophys.Acta_438_437 |
| PubMedID: 952942 |