Sofosbuvir
Sofosbuvir is an orally available nucleotide prodrug and a hepatitis C virus (HCV) NS5B polymerase inhibitor with potential HCV inhibiting activity. Upon oral administration, sofosbuvir is metabolized to 2'-deoxy-2'-alpha-fluoro-beta-C-methyluridine-5'-monophosphate, which is then converted into the active triphosphate nucleotide that inhibits the NS5B polymerase, thereby preventing viral replication.
General
Type : Pro-Drug,Drug,Not A\/B H target
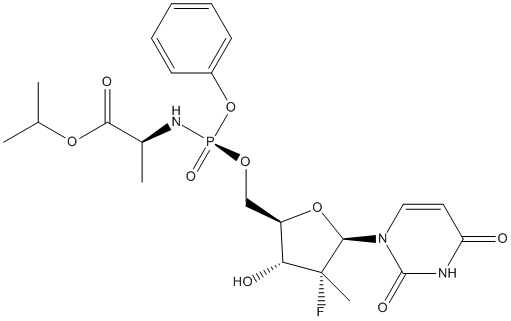
Chemical_Nomenclature : propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate
Canonical SMILES : CC(C)OC(=O)C(C)NP(=O)(OCC1C(C(C(O1)N2C=CC(=O)NC2=O)(C)F)O)OC3=CC=CC=C3
InChI : InChI=1S\/C22H29FN3O9P\/c1-13(2)33-19(29)14(3)25-36(31,35-15-8-6-5-7-9-15)32-12-16-18(28)22(4,23)20(34-16)26-11-10-17(27)24-21(26)30\/h5-11,13-14,16,18,20,28H,12H2,1-4H3,(H,25,31)(H,24,27,30)\/t14-,16+,18+,20+,22+,36-\/m0\/s1
InChIKey : TTZHDVOVKQGIBA-IQWMDFIBSA-N
Other name(s) : PSI-7977,SOVALDI,GS-7977,SCHEMBL2010114,CHEMBL1259059,ZINC100074252,CHEBI:85083,DB08934
MW : 529.5
Formula : C22H29FN3O9P
CAS_number : 1190307-88-0
PubChem : 45375808
UniChem : TTZHDVOVKQGIBA-IQWMDFIBSA-N
IUPHAR :
Wikipedia :
Target
References (4)
| Title : Patient-specific genetic factors predict treatment failure in sofosbuvir-treated patients with chronic hepatitis C - Loucks_2022_Liver.Int_42_796 |
| Author(s) : Loucks CM , Lin JJ , Trueman JN , Drogemoller BI , Wright GEB , Chang WC , Li KH , Yoshida EM , Ford JA , Lee SS , Crotty P , Kim RB , Al-Judaibi B , Schwarz UI , Ramji A , Farivar JF , Tam E , Walston LL , Ross CJD , Carleton BC |
| Ref : Liver Int , 42 :796 , 2022 |
| Abstract : Loucks_2022_Liver.Int_42_796 |
| ESTHER : Loucks_2022_Liver.Int_42_796 |
| PubMedSearch : Loucks_2022_Liver.Int_42_796 |
| PubMedID: 35107877 |
| Title : Covalent CES2 Inhibitors Protect against Reduced Formation of Intestinal Organoids by the Anticancer Drug Irinotecan - Eades_2022_Curr.Drug.Metab__ |
| Author(s) : Eades W , Liu W , Shen Y , Shi Z , Yan B |
| Ref : Curr Drug Metab , : , 2022 |
| Abstract : Eades_2022_Curr.Drug.Metab__ |
| ESTHER : Eades_2022_Curr.Drug.Metab__ |
| PubMedSearch : Eades_2022_Curr.Drug.Metab__ |
| PubMedID: 36515038 |
| Title : Activation of Tenofovir Alafenamide and Sofosbuvir in the Human Lung and Its Implications in the Development of Nucleoside\/Nucleotide Prodrugs for Treating SARS-CoV-2 Pulmonary Infection - Li_2021_Pharmaceutics_13_ |
| Author(s) : Li J , Liu S , Shi J , Zhu HJ |
| Ref : Pharmaceutics , 13 : , 2021 |
| Abstract : Li_2021_Pharmaceutics_13_ |
| ESTHER : Li_2021_Pharmaceutics_13_ |
| PubMedSearch : Li_2021_Pharmaceutics_13_ |
| PubMedID: 34683949 |
| Gene_locus related to this paper: human-CTSA |
| Title : Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977 - Murakami_2010_J.Biol.Chem_285_34337 |
| Author(s) : Murakami E , Tolstykh T , Bao H , Niu C , Steuer HM , Bao D , Chang W , Espiritu C , Bansal S , Lam AM , Otto MJ , Sofia MJ , Furman PA |
| Ref : Journal of Biological Chemistry , 285 :34337 , 2010 |
| Abstract : Murakami_2010_J.Biol.Chem_285_34337 |
| ESTHER : Murakami_2010_J.Biol.Chem_285_34337 |
| PubMedSearch : Murakami_2010_J.Biol.Chem_285_34337 |
| PubMedID: 20801890 |