Imidapril
General
Type : Pro-Drug || Drug || Not A\/B H target
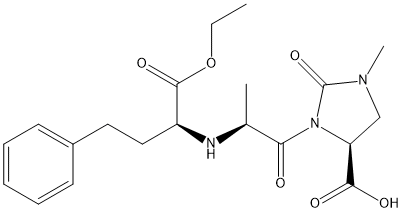
Chemical_Nomenclature : (4S)-3-[(2S)-2-[[(2S)-1-ethoxy-1-oxo-4-phenylbutan-2-yl]amino]propanoyl]-1-methyl-2-oxoimidazolidine-4-carboxylic acid
Canonical SMILES : CCOC(=O)C(CCC1=CC=CC=C1)NC(C)C(=O)N2C(CN(C2=O)C)C(=O)O
InChI : InChI=1S\/C20H27N3O6\/c1-4-29-19(27)15(11-10-14-8-6-5-7-9-14)21-13(2)17(24)23-16(18(25)26)12-22(3)20(23)28\/h5-9,13,15-16,21H,4,10-12H2,1-3H3,(H,25,26)\/t13-,15-,16-\/m0\/s1
InChIKey : KLZWOWYOHUKJIG-BPUTZDHNSA-N
Other name(s) : Imidaprilum, UNII-BW7H1TJS22, Tanatril, CHEMBL317094, SCHEMBL34098, CHEBI:135654, ZINC3784427, DB11783
Target
Families : Carb_B_Chordata
References (11)
| Title : Comparison of substrate specificity among human arylacetamide deacetylase and carboxylesterases - Fukami_2015_Eur.J.Pharm.Sci_78_47 |
| Author(s) : Fukami T , Kariya M , Kurokawa T , Iida A , Nakajima M |
| Ref : Eur J Pharm Sci , 78 :47 , 2015 |
| Abstract : Fukami_2015_Eur.J.Pharm.Sci_78_47 |
| ESTHER : Fukami_2015_Eur.J.Pharm.Sci_78_47 |
| PubMedSearch : Fukami_2015_Eur.J.Pharm.Sci_78_47 |
| PubMedID: 26164127 |
| Gene_locus related to this paper: human-AADAC , human-CES1 , human-CES2 |
| Title : The pharmacogenetics of carboxylesterases: CES1 and CES2 genetic variants and their clinical effect - Merali_2014_Drug.Metabol.Drug.Interact_29_143 |
| Author(s) : Merali Z , Ross S , Pare G |
| Ref : Drug Metabol Drug Interact , 29 :143 , 2014 |
| Abstract : Merali_2014_Drug.Metabol.Drug.Interact_29_143 |
| ESTHER : Merali_2014_Drug.Metabol.Drug.Interact_29_143 |
| PubMedSearch : Merali_2014_Drug.Metabol.Drug.Interact_29_143 |
| PubMedID: 24988246 |
| Gene_locus related to this paper: human-CES2 |
| Title : In Vitro Evaluation of the Inhibitory Potential of Pharmaceutical Excipients on Human Carboxylesterase 1A and 2 - Zhang_2014_PLoS.One_9_e93819 |
| Author(s) : Zhang C , Xu Y , Zhong Q , Li X , Gao P , Feng C , Chu Q , Chen Y , Liu D |
| Ref : PLoS ONE , 9 :e93819 , 2014 |
| Abstract : Zhang_2014_PLoS.One_9_e93819 |
| ESTHER : Zhang_2014_PLoS.One_9_e93819 |
| PubMedSearch : Zhang_2014_PLoS.One_9_e93819 |
| PubMedID: 24699684 |
| Title : Evaluation of the inhibitory effects of antihypertensive drugs on human carboxylesterase in vitro - Yanjiao_2013_Drug.Metab.Pharmacokinet_28_468 |
| Author(s) : Yanjiao X , Chengliang Z , Xiping L , Tao W , Xiuhua R , Dong L |
| Ref : Drug Metab Pharmacokinet , 28 :468 , 2013 |
| Abstract : Yanjiao_2013_Drug.Metab.Pharmacokinet_28_468 |
| ESTHER : Yanjiao_2013_Drug.Metab.Pharmacokinet_28_468 |
| PubMedSearch : Yanjiao_2013_Drug.Metab.Pharmacokinet_28_468 |
| PubMedID: 23648675 |
| Title : In vitro evaluation of inhibitory effects of antidiabetic and antihyperlipidemic drugs on human carboxylesterase activities - Fukami_2010_Drug.Metab.Dispos_38_2173 |
| Author(s) : Fukami T , Takahashi S , Nakagawa N , Maruichi T , Nakajima M , Yokoi T |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 38 :2173 , 2010 |
| Abstract : Fukami_2010_Drug.Metab.Dispos_38_2173 |
| ESTHER : Fukami_2010_Drug.Metab.Dispos_38_2173 |
| PubMedSearch : Fukami_2010_Drug.Metab.Dispos_38_2173 |
| PubMedID: 20810539 |
| Title : Different inhibitory effects in rat and human carboxylesterases - Takahashi_2009_Drug.Metab.Dispos_37_956 |
| Author(s) : Takahashi S , Katoh M , Saitoh T , Nakajima M , Yokoi T |
| Ref : Drug Metabolism & Disposition: The Biological Fate of Chemicals , 37 :956 , 2009 |
| Abstract : Takahashi_2009_Drug.Metab.Dispos_37_956 |
| ESTHER : Takahashi_2009_Drug.Metab.Dispos_37_956 |
| PubMedSearch : Takahashi_2009_Drug.Metab.Dispos_37_956 |
| PubMedID: 19225040 |
| Title : Functional polymorphisms in carboxylesterase1A2 (CES1A2) gene involves specific protein 1 (Sp1) binding sites - Yoshimura_2008_Biochem.Biophys.Res.Commun_369_939 |
| Author(s) : Yoshimura M , Kimura T , Ishii M , Ishii K , Matsuura T , Geshi E , Hosokawa M , Muramatsu M |
| Ref : Biochemical & Biophysical Research Communications , 369 :939 , 2008 |
| Abstract : Yoshimura_2008_Biochem.Biophys.Res.Commun_369_939 |
| ESTHER : Yoshimura_2008_Biochem.Biophys.Res.Commun_369_939 |
| PubMedSearch : Yoshimura_2008_Biochem.Biophys.Res.Commun_369_939 |
| PubMedID: 18328811 |
| Gene_locus related to this paper: human-CES1 |
| Title : Allosteric kinetics of human carboxylesterase 1: species differences and interindividual variability - Takahashi_2008_J.Pharm.Sci_97_5434 |
| Author(s) : Takahashi S , Katoh M , Saitoh T , Nakajima M , Yokoi T |
| Ref : J Pharm Sci , 97 :5434 , 2008 |
| Abstract : Takahashi_2008_J.Pharm.Sci_97_5434 |
| ESTHER : Takahashi_2008_J.Pharm.Sci_97_5434 |
| PubMedSearch : Takahashi_2008_J.Pharm.Sci_97_5434 |
| PubMedID: 18383336 |
| Title : Involvement of human blood arylesterases and liver microsomal carboxylesterases in nafamostat hydrolysis - Yamaori_2006_Drug.Metab.Pharmacokinet_21_147 |
| Author(s) : Yamaori S , Fujiyama N , Kushihara M , Funahashi T , Kimura T , Yamamoto I , Sone T , Isobe M , Ohshima T , Matsumura K , Oda M , Watanabe K |
| Ref : Drug Metab Pharmacokinet , 21 :147 , 2006 |
| Abstract : Yamaori_2006_Drug.Metab.Pharmacokinet_21_147 |
| ESTHER : Yamaori_2006_Drug.Metab.Pharmacokinet_21_147 |
| PubMedSearch : Yamaori_2006_Drug.Metab.Pharmacokinet_21_147 |
| PubMedID: 16702735 |
| Title : A single nucleotide polymorphism in the carboxylesterase gene is associated with the responsiveness to imidapril medication and the promoter activity - Geshi_2005_Hypertens.Res_28_719 |
| Author(s) : Geshi E , Kimura T , Yoshimura M , Suzuki H , Koba S , Sakai T , Saito T , Koga A , Muramatsu M , Katagiri T |
| Ref : Hypertens Res , 28 :719 , 2005 |
| Abstract : Geshi_2005_Hypertens.Res_28_719 |
| ESTHER : Geshi_2005_Hypertens.Res_28_719 |
| PubMedSearch : Geshi_2005_Hypertens.Res_28_719 |
| PubMedID: 16419644 |
| Gene_locus related to this paper: human-CES1 |
| Title : Hydrolytic profile for ester- or amide-linkage by carboxylesterases pI 5.3 and 4.5 from human liver - Takai_1997_Biol.Pharm.Bull_20_869 |
| Author(s) : Takai S , Matsuda A , Usami Y , Adachi T , Sugiyama T , Katagiri Y , Tatematsu M , Hirano K |
| Ref : Biol Pharm Bull , 20 :869 , 1997 |
| Abstract : Takai_1997_Biol.Pharm.Bull_20_869 |
| ESTHER : Takai_1997_Biol.Pharm.Bull_20_869 |
| PubMedSearch : Takai_1997_Biol.Pharm.Bull_20_869 |
| PubMedID: 9300133 |