WWL50
Carbamate inhibitor of mouse CES1 The fluoride compound is WWL50, without fluoride CHEMBL77270 CID 306692
General
Type : Carbamate
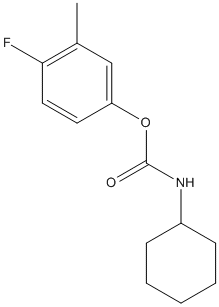
Chemical_Nomenclature : 4-fluoro-3-methylphenyl cyclohexylcarbamate || (3-methylphenyl) N-cyclohexylcarbamate
Canonical SMILES : CC1=CC(=CC=C1)OC(=O)NC2CCCCC2
InChI : InChI=1S\/C14H19NO2\/c1-11-6-5-9-13(10-11)17-14(16)15-12-7-3-2-4-8-12\/h5-6,9-10,12H,2-4,7-8H2,1H3,(H,15,16)
InChIKey : HVJIDJLSBZMPDP-UHFFFAOYSA-N
Other name(s) : CHEMBL77270 || NSC204378 || ZINC1738990 || BDBM50128584 || Cyclohexyl-carbamic acid m-tolyl ester
MW : 251.30
Formula : C14H18FNO2
CAS_number :
PubChem : 306692
UniChem : HVJIDJLSBZMPDP-UHFFFAOYSA-N
Target
Families : WWL50 ligand of proteins in family
Carb_B_Chordata
References (3)
| Title : Substrate-Competitive Activity-Based Profiling of Ester Prodrug Activating Enzymes - Xu_2015_Mol.Pharm_12_3399 |
| Author(s) : Xu H , Majmudar JD , Davda D , Ghanakota P , Kim KH , Carlson HA , Showalter HD , Martin BR , Amidon GL |
| Ref : Mol Pharm , 12 :3399 , 2015 |
| Abstract : |
| PubMedSearch : Xu_2015_Mol.Pharm_12_3399 |
| PubMedID: 26262434 |
| Gene_locus related to this paper: human-CES1 |
| Title : Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening - Bachovchin_2010_Proc.Natl.Acad.Sci.U.S.A_107_20941 |
| Author(s) : Bachovchin DA , Ji T , Li W , Simon GM , Blankman JL , Adibekian A , Hoover H , Niessen S , Cravatt BF |
| Ref : Proc Natl Acad Sci U S A , 107 :20941 , 2010 |
| Abstract : |
| PubMedSearch : Bachovchin_2010_Proc.Natl.Acad.Sci.U.S.A_107_20941 |
| PubMedID: 21084632 |
| Title : Design, synthesis, and structure-activity relationships of alkylcarbamic acid aryl esters, a new class of fatty acid amide hydrolase inhibitors - Tarzia_2003_J.Med.Chem_46_2352 |
| Author(s) : Tarzia G , Duranti A , Tontini A , Piersanti G , Mor M , Rivara S , Plazzi PV , Park C , Kathuria S , Piomelli D |
| Ref : Journal of Medicinal Chemistry , 46 :2352 , 2003 |
| Abstract : |
| PubMedSearch : Tarzia_2003_J.Med.Chem_46_2352 |
| PubMedID: 12773040 |