Edrophonium
A rapid-onset, short-acting cholinesterase inhibitor used in cardiac arrhythmias and in the diagnosis of myasthenia gravis. It has also been used as an antidote to curare principles
General
Type : Drug,Quaternary compound
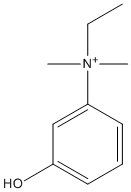
Chemical_Nomenclature : ethyl-(3-hydroxyphenyl)-dimethylazanium
Canonical SMILES : CC[N+](C)(C)C1=CC(=CC=C1)O
InChI : InChI=1S\/C10H15NO\/c1-4-11(2,3)9-6-5-7-10(12)8-9\/h5-8H,4H2,1-3H3\/p+1
InChIKey : VWLHWLSRQJQWRG-UHFFFAOYSA-O
Other name(s) : Tensilon,R02-3198,Edrophonum,Reversol,Edroponium,Enlon,Ethyl (m-hydroxyphenyl) dimethylammonium bromide,(3-hydroxyphenyl) dimethylethylammonium bromide,dimethylethyl (3-hydroxyphenyl) ammonium bromide,3-hydroxy-N,N-dimethyl-N-ethylanilinium bromide,EDR
MW : 201.69
Formula : C10H16ClNO
CAS_number : 116-38-1
PubChem : 3202
UniChem : VWLHWLSRQJQWRG-UHFFFAOYSA-O
IUPHAR : 9073
Wikipedia : Edrophonium
Target
Families : Edrophonium ligand of proteins in family: ACHE || BCHE
Stucture :
Protein : torca-ACHE || human-ACHE
References (35)
| Title : beta-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies - Bartolini_2003_Biochem.Pharmacol_65_407 |
| Author(s) : Bartolini M , Bertucci C , Cavrini V , Andrisano V |
| Ref : Biochemical Pharmacology , 65 :407 , 2003 |
| Abstract : Bartolini_2003_Biochem.Pharmacol_65_407 |
| ESTHER : Bartolini_2003_Biochem.Pharmacol_65_407 |
| PubMedSearch : Bartolini_2003_Biochem.Pharmacol_65_407 |
| PubMedID: 12527333 |
| Title : Effect of tetramethylammonium, choline and edrophonium on insect acetylcholinesterase: test of a kinetic model - Stojan_1999_Chem.Biol.Interact_119-120_137 |
| Author(s) : Stojan J , Marcel V , Fournier D |
| Ref : Chemico-Biological Interactions , 119-120 :137 , 1999 |
| Abstract : Stojan_1999_Chem.Biol.Interact_119-120_137 |
| ESTHER : Stojan_1999_Chem.Biol.Interact_119-120_137 |
| PubMedSearch : Stojan_1999_Chem.Biol.Interact_119-120_137 |
| PubMedID: 10421447 |
| Title : Effects of four anticholinesterase-anticholinergic combinations and tracheal extubation on QTc interval of the ECG, heart rate and arterial pressure - Saarnivaara_1998_Acta.Anaesthesiol.Scand_42_460 |
| Author(s) : Saarnivaara L , Simola M |
| Ref : Acta Anaesthesiologica Scandinavica , 42 :460 , 1998 |
| Abstract : Saarnivaara_1998_Acta.Anaesthesiol.Scand_42_460 |
| ESTHER : Saarnivaara_1998_Acta.Anaesthesiol.Scand_42_460 |
| PubMedSearch : Saarnivaara_1998_Acta.Anaesthesiol.Scand_42_460 |
| PubMedID: 9563867 |
| Title : Comparison of tilt angles and provocative agents (edrophonium and isoproterenol) to improve head-upright tilt-table testing - Voice_1998_Am.J.Cardiol_81_346 |
| Author(s) : Voice RA , Lurie KG , Sakaguchi S , Rector TS , Benditt DG |
| Ref : American Journal of Cardiology , 81 :346 , 1998 |
| Abstract : Voice_1998_Am.J.Cardiol_81_346 |
| ESTHER : Voice_1998_Am.J.Cardiol_81_346 |
| PubMedSearch : Voice_1998_Am.J.Cardiol_81_346 |
| PubMedID: 9468082 |
| Title : Inhibition of cholinesterase-associated aryl acylamidase activity by anticholinesterase agents: focus on drugs potentially effective in Alzheimer's disease - Costagli_1998_Biochem.Pharmacol_55_1733 |
| Author(s) : Costagli C , Galli A |
| Ref : Biochemical Pharmacology , 55 :1733 , 1998 |
| Abstract : Costagli_1998_Biochem.Pharmacol_55_1733 |
| ESTHER : Costagli_1998_Biochem.Pharmacol_55_1733 |
| PubMedSearch : Costagli_1998_Biochem.Pharmacol_55_1733 |
| PubMedID: 9634011 |
| Title : Static laue diffraction studies on acetylcholinesterase - Ravelli_1998_Acta.Crystallogr.D.Biol.Crystallogr_54_1359 |
| Author(s) : Ravelli RB , Raves ML , Ren Z , Bourgeois D , Roth M , Kroon J , Silman I , Sussman JL |
| Ref : Acta Crystallographica D Biol Crystallogr , 54 :1359 , 1998 |
| Abstract : Ravelli_1998_Acta.Crystallogr.D.Biol.Crystallogr_54_1359 |
| ESTHER : Ravelli_1998_Acta.Crystallogr.D.Biol.Crystallogr_54_1359 |
| PubMedSearch : Ravelli_1998_Acta.Crystallogr.D.Biol.Crystallogr_54_1359 |
| PubMedID: 10089512 |
| Gene_locus related to this paper: torca-ACHE |
| Title : Neostigmine but not edrophonium prolongs the action of mivacurium - Symington_1996_Can.J.Anaesth_43_1220 |
| Author(s) : Symington MJ , Mirakhur RK , Kumar N |
| Ref : Canadian Journal of Anaesthesia , 43 :1220 , 1996 |
| Abstract : Symington_1996_Can.J.Anaesth_43_1220 |
| ESTHER : Symington_1996_Can.J.Anaesth_43_1220 |
| PubMedSearch : Symington_1996_Can.J.Anaesth_43_1220 |
| PubMedID: 8955970 |
| Title : Cloning and expression of acetylcholinesterase from Bungarus fasciatus venom. A new type of cooh-terminal domain\; involvement of a positively charged residue in the peripheral site - Cousin_1996_J.Biol.Chem_271_15099 |
| Author(s) : Cousin X , Bon S , Duval N , Massoulie J , Bon C |
| Ref : Journal of Biological Chemistry , 271 :15099 , 1996 |
| Abstract : Cousin_1996_J.Biol.Chem_271_15099 |
| ESTHER : Cousin_1996_J.Biol.Chem_271_15099 |
| PubMedSearch : Cousin_1996_J.Biol.Chem_271_15099 |
| PubMedID: 8662867 |
| Gene_locus related to this paper: bunfa-ACHE |
| Title : Dose responses for neostigmine and edrophonium as antagonists of mivacurium in adults and children - Bevan_1996_Anesthesiology_84_354 |
| Author(s) : Bevan JC , Tousignant C , Stephenson C , Blackman L , Reimer E , Smith MF , Bevan DR |
| Ref : Anesthesiology , 84 :354 , 1996 |
| Abstract : Bevan_1996_Anesthesiology_84_354 |
| ESTHER : Bevan_1996_Anesthesiology_84_354 |
| PubMedSearch : Bevan_1996_Anesthesiology_84_354 |
| PubMedID: 8602666 |
| Title : Anticholinesterase drugs stimulate phosphatidylinositol response in rat tracheal slices - Shibata_1996_Anesth.Analg_82_1211 |
| Author(s) : Shibata O , Kanairo M , Zhang S , Hasuo H , Morooka H , Fujie T , Sumikawa K |
| Ref : Anesthesia & Analgesia , 82 :1211 , 1996 |
| Abstract : Shibata_1996_Anesth.Analg_82_1211 |
| ESTHER : Shibata_1996_Anesth.Analg_82_1211 |
| PubMedSearch : Shibata_1996_Anesth.Analg_82_1211 |
| PubMedID: 8638793 |
| Title : Different properties of the bradycardia produced by neostigmine and edrophonium in the cat - Backman_1996_Can.J.Anaesth_43_731 |
| Author(s) : Backman SB , Stein RD , Blank DW , Collier B , Polosa C |
| Ref : Canadian Journal of Anaesthesia , 43 :731 , 1996 |
| Abstract : Backman_1996_Can.J.Anaesth_43_731 |
| ESTHER : Backman_1996_Can.J.Anaesth_43_731 |
| PubMedSearch : Backman_1996_Can.J.Anaesth_43_731 |
| PubMedID: 8807181 |
| Title : Acetylcholinesterase in Dendrobaena veneta (Oligochaeta: Opisthopora) is present with forms sensitive and insensitive to phosphatidylinositol phospholipase C. Biochemical characterization and histochemical localization in the nervous system - Talesa_1996_Eur.J.Biochem_238_538 |
| Author(s) : Talesa V , Romani R , Rosi G , Giovannini E |
| Ref : European Journal of Biochemistry , 238 :538 , 1996 |
| Abstract : Talesa_1996_Eur.J.Biochem_238_538 |
| ESTHER : Talesa_1996_Eur.J.Biochem_238_538 |
| PubMedSearch : Talesa_1996_Eur.J.Biochem_238_538 |
| PubMedID: 8681969 |
| Title : Comparative pharmacokinetics of four cholinesterase inhibitors in rats - Yamamoto_1995_Biol.Pharm.Bull_18_1292 |
| Author(s) : Yamamoto K , Sawada Y , Iga T |
| Ref : Biol Pharm Bull , 18 :1292 , 1995 |
| Abstract : Yamamoto_1995_Biol.Pharm.Bull_18_1292 |
| ESTHER : Yamamoto_1995_Biol.Pharm.Bull_18_1292 |
| PubMedSearch : Yamamoto_1995_Biol.Pharm.Bull_18_1292 |
| PubMedID: 8845827 |
| Title : Contribution of aromatic moieties of tyrosine 133 and of the anionic subsite tryptophan 86 to catalytic efficiency and allosteric modulation of acetylcholinesterase - Ordentlich_1995_J.Biol.Chem_270_2082 |
| Author(s) : Ordentlich A , Barak D , Kronman C , Ariel N , Segall Y , Velan B , Shafferman A |
| Ref : Journal of Biological Chemistry , 270 :2082 , 1995 |
| Abstract : Ordentlich_1995_J.Biol.Chem_270_2082 |
| ESTHER : Ordentlich_1995_J.Biol.Chem_270_2082 |
| PubMedSearch : Ordentlich_1995_J.Biol.Chem_270_2082 |
| PubMedID: 7836436 |
| Title : Solubilization, molecular forms, purification and substrate specificity of two acetylcholinesterases in the medicinal leech (Hirudo medicinalis) - Talesa_1995_Biochem.J_306_687 |
| Author(s) : Talesa V , Grauso M , Giovannini E , Rosi G , Toutant JP |
| Ref : Biochemical Journal , 306 :687 , 1995 |
| Abstract : Talesa_1995_Biochem.J_306_687 |
| ESTHER : Talesa_1995_Biochem.J_306_687 |
| PubMedSearch : Talesa_1995_Biochem.J_306_687 |
| PubMedID: 7702560 |
| Title : Fasciculin 2 binds to the peripheral site on acetylcholinesterase and inhibits substrate hydrolysis by slowing a step involving proton transfer during enzyme acylation - Eastman_1995_J.Biol.Chem_270_19694 |
| Author(s) : Eastman J , Wilson EJ , Cervenansky C , Rosenberry TL |
| Ref : Journal of Biological Chemistry , 270 :19694 , 1995 |
| Abstract : Eastman_1995_J.Biol.Chem_270_19694 |
| ESTHER : Eastman_1995_J.Biol.Chem_270_19694 |
| PubMedSearch : Eastman_1995_J.Biol.Chem_270_19694 |
| PubMedID: 7649979 |
| Title : Comparison of the effects of neostigmine and edrophonium on the duration of action of suxamethonium - McCoy_1995_Acta.Anaesthesiol.Scand_39_744 |
| Author(s) : McCoy EP , Mirakhur RK |
| Ref : Acta Anaesthesiologica Scandinavica , 39 :744 , 1995 |
| Abstract : McCoy_1995_Acta.Anaesthesiol.Scand_39_744 |
| ESTHER : McCoy_1995_Acta.Anaesthesiol.Scand_39_744 |
| PubMedSearch : McCoy_1995_Acta.Anaesthesiol.Scand_39_744 |
| PubMedID: 7484027 |
| Title : 6-Coumarin diazonium salt: a specific affinity label of the Torpedo acetylcholinesterase peripheral site - Schalk_1995_Mol.Pharmacol_48_1063 |
| Author(s) : Schalk I , Ehret-Sabatier L , Le Feuvre Y , Bon S , Massoulie J , Goeldner M |
| Ref : Molecular Pharmacology , 48 :1063 , 1995 |
| Abstract : Schalk_1995_Mol.Pharmacol_48_1063 |
| ESTHER : Schalk_1995_Mol.Pharmacol_48_1063 |
| PubMedSearch : Schalk_1995_Mol.Pharmacol_48_1063 |
| PubMedID: 8848006 |
| Title : Edrophonium increases mivacurium concentrations during constant mivacurium infusion, and large doses minimally antagonize paralysis [see comments] - Hart_1995_Anesthesiology_82_912 |
| Author(s) : Hart PS , Wright PM , Brown R , Lau M , Sharma ML , Miller RD , Gruenke L , Fisher DM |
| Ref : Anesthesiology , 82 :912 , 1995 |
| Abstract : Hart_1995_Anesthesiology_82_912 |
| ESTHER : Hart_1995_Anesthesiology_82_912 |
| PubMedSearch : Hart_1995_Anesthesiology_82_912 |
| PubMedID: 7717563 |
| Title : Differential effects of peripheral site ligands on Torpedo and chicken acetylcholinesterase - Eichler_1994_Mol.Pharmacol_45_335 |
| Author(s) : Eichler J , Anselmet A , Sussman JL , Massoulie J , Silman I |
| Ref : Molecular Pharmacology , 45 :335 , 1994 |
| Abstract : Eichler_1994_Mol.Pharmacol_45_335 |
| ESTHER : Eichler_1994_Mol.Pharmacol_45_335 |
| PubMedSearch : Eichler_1994_Mol.Pharmacol_45_335 |
| PubMedID: 8114681 |
| Title : Acetylcholinesterase peripheral anionic site degeneracy conferred by amino acid arrays sharing a common core - Barak_1994_J.Biol.Chem_269_6296 |
| Author(s) : Barak D , Kronman C , Ordentlich A , Ariel N , Bromberg A , Marcus D , Lazar A , Velan B , Shafferman A |
| Ref : Journal of Biological Chemistry , 269 :6296 , 1994 |
| Abstract : Barak_1994_J.Biol.Chem_269_6296 |
| ESTHER : Barak_1994_J.Biol.Chem_269_6296 |
| PubMedSearch : Barak_1994_J.Biol.Chem_269_6296 |
| PubMedID: 8119978 |
| Title : The back door hypothesis for product clearance in acetylcholinesterase challenged by site-directed mutagenesis - Kronman_1994_J.Biol.Chem_269_27819 |
| Author(s) : Kronman C , Ordentlich A , Barak D , Velan B , Shafferman A |
| Ref : Journal of Biological Chemistry , 269 :27819 , 1994 |
| Abstract : Kronman_1994_J.Biol.Chem_269_27819 |
| ESTHER : Kronman_1994_J.Biol.Chem_269_27819 |
| PubMedSearch : Kronman_1994_J.Biol.Chem_269_27819 |
| PubMedID: 7961709 |
| Title : Electrostatic attraction by surface charge does not contribute to the catalytic efficiency of acetylcholinesterase - Shafferman_1994_EMBO.J_13_3448 |
| Author(s) : Shafferman A , Ordentlich A , Barak D , Kronman C , Ber R , Bino T , Ariel N , Osman R , Velan B |
| Ref : EMBO Journal , 13 :3448 , 1994 |
| Abstract : Shafferman_1994_EMBO.J_13_3448 |
| ESTHER : Shafferman_1994_EMBO.J_13_3448 |
| PubMedSearch : Shafferman_1994_EMBO.J_13_3448 |
| PubMedID: 8062821 |
| Title : The molecular forms of acetylcholinesterase from Necator americanus (Nematoda), a hookworm parasite of the human intestine - Pritchard_1994_Eur.J.Biochem_219_317 |
| Author(s) : Pritchard DI , Brown A , Toutant JP |
| Ref : European Journal of Biochemistry , 219 :317 , 1994 |
| Abstract : Pritchard_1994_Eur.J.Biochem_219_317 |
| ESTHER : Pritchard_1994_Eur.J.Biochem_219_317 |
| PubMedSearch : Pritchard_1994_Eur.J.Biochem_219_317 |
| PubMedID: 8306998 |
| Title : Fresh frozen plasma and edrophonium in a patient with a plasma cholinesterase deficiency - Smith_1993_Anaesthesia_48_511 |
| Author(s) : Smith DC , Ridley SA , Donaldson KF |
| Ref : Anaesthesia , 48 :511 , 1993 |
| Abstract : Smith_1993_Anaesthesia_48_511 |
| ESTHER : Smith_1993_Anaesthesia_48_511 |
| PubMedSearch : Smith_1993_Anaesthesia_48_511 |
| PubMedID: 8391759 |
| Title : Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors - Radic_1993_Biochemistry_32_12074 |
| Author(s) : Radic Z , Pickering NA , Vellom DC , Camp S , Taylor P |
| Ref : Biochemistry , 32 :12074 , 1993 |
| Abstract : Radic_1993_Biochemistry_32_12074 |
| ESTHER : Radic_1993_Biochemistry_32_12074 |
| PubMedSearch : Radic_1993_Biochemistry_32_12074 |
| PubMedID: 8218285 |
| Title : Dissection of the human acetylcholinesterase active center determinants of substrate specificity. Identification of residues constituting the anionic site, the hydrophobic site, and the acyl pocket - Ordentlich_1993_J.Biol.Chem_268_17083 |
| Author(s) : Ordentlich A , Barak D , Kronman C , Flashner Y , Leitner M , Segall Y , Ariel N , Cohen S , Velan B , Shafferman A |
| Ref : Journal of Biological Chemistry , 268 :17083 , 1993 |
| Abstract : Ordentlich_1993_J.Biol.Chem_268_17083 |
| ESTHER : Ordentlich_1993_J.Biol.Chem_268_17083 |
| PubMedSearch : Ordentlich_1993_J.Biol.Chem_268_17083 |
| PubMedID: 8349597 |
| Gene_locus related to this paper: human-ACHE , human-BCHE |
| Title : Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase - Harel_1993_Proc.Natl.Acad.Sci.U.S.A_90_9031 |
| Author(s) : Harel M , Schalk I , Ehret-Sabatier L , Bouet F , Goeldner M , Hirth C , Axelsen PH , Silman I , Sussman JL |
| Ref : Proceedings of the National Academy of Sciences of the United States of America , 90 :9031 , 1993 |
| Abstract : Harel_1993_Proc.Natl.Acad.Sci.U.S.A_90_9031 |
| ESTHER : Harel_1993_Proc.Natl.Acad.Sci.U.S.A_90_9031 |
| PubMedSearch : Harel_1993_Proc.Natl.Acad.Sci.U.S.A_90_9031 |
| PubMedID: 8415649 |
| Gene_locus related to this paper: torca-ACHE |
| Title : Substrate inhibition of acetylcholinesterase: residues affecting signal transduction from the surface to the catalytic center - Shafferman_1992_EMBO.J_11_3561 |
| Author(s) : Shafferman A , Velan B , Ordentlich A , Kronman C , Grosfeld H , Leitner M , Flashner Y , Cohen S , Barak D , Ariel N |
| Ref : EMBO Journal , 11 :3561 , 1992 |
| Abstract : Shafferman_1992_EMBO.J_11_3561 |
| ESTHER : Shafferman_1992_EMBO.J_11_3561 |
| PubMedSearch : Shafferman_1992_EMBO.J_11_3561 |
| PubMedID: 1396557 |
| Title : The effect of the organophosphate trichlorfon on the neuromuscular blocking activity of atracurium in halothane-anesthetized horses - Hildebrand_1989_J.Vet.Pharmacol.Ther_12_277 |
| Author(s) : Hildebrand SV , Hill Td , Holland M |
| Ref : J Vet Pharmacol Ther , 12 :277 , 1989 |
| Abstract : Hildebrand_1989_J.Vet.Pharmacol.Ther_12_277 |
| ESTHER : Hildebrand_1989_J.Vet.Pharmacol.Ther_12_277 |
| PubMedSearch : Hildebrand_1989_J.Vet.Pharmacol.Ther_12_277 |
| PubMedID: 2810476 |
| Title : Ligand stabilization of cholinesterases - Payne_1989_Biochim.Biophys.Acta_999_46 |
| Author(s) : Payne CS , Saeed M , Wolfe AD |
| Ref : Biochimica & Biophysica Acta , 999 :46 , 1989 |
| Abstract : Payne_1989_Biochim.Biophys.Acta_999_46 |
| ESTHER : Payne_1989_Biochim.Biophys.Acta_999_46 |
| PubMedSearch : Payne_1989_Biochim.Biophys.Acta_999_46 |
| PubMedID: 2804138 |
| Title : Structure-activity relationship of reversible cholinesterase inhibitors including paraquat - Seto_1988_Arch.Toxicol_62_37 |
| Author(s) : Seto Y , Shinohara T |
| Ref : Archives of Toxicology , 62 :37 , 1988 |
| Abstract : Seto_1988_Arch.Toxicol_62_37 |
| ESTHER : Seto_1988_Arch.Toxicol_62_37 |
| PubMedSearch : Seto_1988_Arch.Toxicol_62_37 |
| PubMedID: 3190453 |
| Title : Inhibition of human motor endplate cholinesterase by anticholinesterase compounds - Fujii_1982_Acta.Med.Okayama_36_229 |
| Author(s) : Fujii M , Namba T |
| Ref : Acta Med Okayama , 36 :229 , 1982 |
| Abstract : Fujii_1982_Acta.Med.Okayama_36_229 |
| ESTHER : Fujii_1982_Acta.Med.Okayama_36_229 |
| PubMedSearch : Fujii_1982_Acta.Med.Okayama_36_229 |
| PubMedID: 7113748 |
| Title : Interaction of fluorescence probes with acetylcholinesterase. The site and specificity of propidium binding - Taylor_1975_Biochemistry_14_1989 |
| Author(s) : Taylor P , Lappi S |
| Ref : Biochemistry , 14 :1989 , 1975 |
| Abstract : Taylor_1975_Biochemistry_14_1989 |
| ESTHER : Taylor_1975_Biochemistry_14_1989 |
| PubMedSearch : Taylor_1975_Biochemistry_14_1989 |
| PubMedID: 1125207 |