CBDP
cyclic metabolite of tri-o-cresyl phosphate; synergist for cholinesterase inhibitor, carboxylesterase inhibitor, close to phenylsaligenin cyclic phosphate. Saligenin cyclic o-tolyl phosphate (SCOTP) has been proposed as the active metabolite of tri-o-cresyl phosphate (TOCP), a neurotoxic organophosphate. TRI-O-CRESYL PHOSPHATE (TOCP) is metabolized in vitro and in vivo to form potent esterase inhibitors. CBDP and Phenyl-OBDP are potent NTE inhibitors. TRI-O-CBESYL PHOSPHATE (TOCP) is metabolized in vitro and in vivo to form potent esterase inhibitors. CBDP and Phenyl-BDPO are potent NTE inhibitors
General
Type : Organophosphate,Cyclic OP,NTE inhibitor,Not A\/B H target
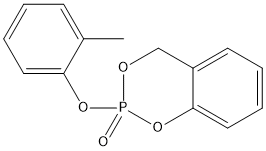
Chemical_Nomenclature : 2-(2-methylphenoxy)-4H-1,3,2$l^{5}-benzodioxaphosphinine 2-oxide
Canonical SMILES : CC1=CC=CC=C1OP2(=O)OCC3=CC=CC=C3O2
InChI : InChI=1S\/C14H13O4P\/c1-11-6-2-4-8-13(11)17-19(15)16-10-12-7-3-5-9-14(12)18-19\/h2-9H,10H2,1H3
InChIKey : LKQSEFCGKYFESN-UHFFFAOYSA-N
Other name(s) : BRN 2538084,2-(o-Cresyl)-4H-1,3,2-benzodioxaphoran-2-one,2-(2-Cresyl)-4H-1-3-2-benzodioxaphosphorin-2-oxide,4H-1,3,2-Benzodioxaphosphorin, 2-(o-tolyloxy)-, 2-oxide,4H-1,3,2-Benzodioxaphosphorin, 2-(2-methylphenoxy)-, 2-oxide,2-(o-cresyl)-4H-1,3,2-benzodioxaphosphoran-2-one,2-(2-cresyl)-4H-1,3,2-benzodioxaphosphorin-2-oxide,LS-107599,2-tolylsaligenin phosphate,cresylsaligenyl phosphate,cresylsaligenin phosphate,2-TSLPH,o-TSP,saligenin cyclic-3-tolylphosphate,SCOTP,0-tosylsaligenin phosphate,2-(o-cresyl)-4H-1-3-2-benzodioxaphosphorin-2-oxide,C000516,2-(o-cresyl)-4H-1:3:2-benzodioxaphosphorin-2-oxide
MW : 276
Formula : C14H13O4P
CAS_number : 1222-87-3
PubChem : 104993
UniChem : LKQSEFCGKYFESN-UHFFFAOYSA-N
IUPHAR :
Wikipedia :
Target
Families : CBDP ligand of proteins in family: ACHE || BCHE || Carb_B_Chordata
Stucture :
Protein : human-BCHE || mouse-ACHE
References (42)
| Title : Inhibition kinetics of acetylcholinesterase and butyrylcholinesterase from various species by 2-(2-cresyl)-4H-1,3,2-benzodioxaphosphorin-2-oxide (CBDP) - Horn_2024_Toxicol.Lett_396_28 |
| Author(s) : Horn G , Rappengluck S , Worek F |
| Ref : Toxicol Lett , 396 :28 , 2024 |
| Abstract : Horn_2024_Toxicol.Lett_396_28 |
| ESTHER : Horn_2024_Toxicol.Lett_396_28 |
| PubMedSearch : Horn_2024_Toxicol.Lett_396_28 |
| PubMedID: 38642675 |
| Title : 1-(3-Tert-Butylphenyl)-2,2,2-Trifluoroethanone as a Potent Transition-State Analogue Slow-Binding Inhibitor of Human Acetylcholinesterase: Kinetic, MD and QM\/MM Studies - Zueva_2020_Biomolecules_10_1608 |
| Author(s) : Zueva IV , Lushchekina SV , Pottie IR , Darvesh S , Masson P |
| Ref : Biomolecules , 10 : , 2020 |
| Abstract : Zueva_2020_Biomolecules_10_1608 |
| ESTHER : Zueva_2020_Biomolecules_10_1608 |
| PubMedSearch : Zueva_2020_Biomolecules_10_1608 |
| PubMedID: 33260981 |
| Title : Assessment of neurotoxic effects of tri-cresyl phosphates (TCPs) and cresyl saligenin phosphate (CBDP) using a combination of in vitro techniques - Hausherr_2017_Neurotoxicol_59_210 |
| Author(s) : Hausherr V , Schobel N , Liebing J , van Thriel C |
| Ref : Neurotoxicology , 59 :210 , 2017 |
| Abstract : Hausherr_2017_Neurotoxicol_59_210 |
| ESTHER : Hausherr_2017_Neurotoxicol_59_210 |
| PubMedSearch : Hausherr_2017_Neurotoxicol_59_210 |
| PubMedID: 27288108 |
| Title : Saligenin cyclic-o-tolyl phosphate (SCOTP) induces autophagy of rat spermatogonial stem cells - Xu_2016_Reprod.Toxicol_60_62 |
| Author(s) : Xu LL , Liu ML , Wang JL , Yu M , Chen JX |
| Ref : Reprod Toxicol , 60 :62 , 2016 |
| Abstract : Xu_2016_Reprod.Toxicol_60_62 |
| ESTHER : Xu_2016_Reprod.Toxicol_60_62 |
| PubMedSearch : Xu_2016_Reprod.Toxicol_60_62 |
| PubMedID: 26815770 |
| Title : The effects of 8-OH-DPAT on neuroinflammation after sarin exposure in mice - Garrett_2013_Toxicology_310_22 |
| Author(s) : Garrett TL , Joshi K , Rapp CM , Chapleau M , Cool DR , Schlager JJ , Lucot JB |
| Ref : Toxicology , 310 :22 , 2013 |
| Abstract : Garrett_2013_Toxicology_310_22 |
| ESTHER : Garrett_2013_Toxicology_310_22 |
| PubMedSearch : Garrett_2013_Toxicology_310_22 |
| PubMedID: 23692952 |
| Title : Cresyl saligenin phosphate makes multiple adducts on free histidine, but does not form an adduct on histidine 438 of human butyrylcholinesterase - Liyasova_2013_Chem.Biol.Interact_203_103 |
| Author(s) : Liyasova MS , Schopfer LM , Lockridge O |
| Ref : Chemico-Biological Interactions , 203 :103 , 2013 |
| Abstract : Liyasova_2013_Chem.Biol.Interact_203_103 |
| ESTHER : Liyasova_2013_Chem.Biol.Interact_203_103 |
| PubMedSearch : Liyasova_2013_Chem.Biol.Interact_203_103 |
| PubMedID: 22898212 |
| Title : Inhibition pathways of the potent organophosphate CBDP with cholinesterases revealed by X-ray crystallographic snapshots and mass spectrometry - Carletti_2013_Chem.Res.Toxicol_26_280 |
| Author(s) : Carletti E , Colletier JP , Schopfer LM , Santoni G , Masson P , Lockridge O , Nachon F , Weik M |
| Ref : Chemical Research in Toxicology , 26 :280 , 2013 |
| Abstract : Carletti_2013_Chem.Res.Toxicol_26_280 |
| ESTHER : Carletti_2013_Chem.Res.Toxicol_26_280 |
| PubMedSearch : Carletti_2013_Chem.Res.Toxicol_26_280 |
| PubMedID: 23339663 |
| Gene_locus related to this paper: human-BCHE , mouse-ACHE |
| Title : Effects of viscosity and osmotic stress on the reaction of human butyrylcholinesterase with cresyl saligenin phosphate, a toxicant related to aerotoxic syndrome: kinetic and molecular dynamics studies - Masson_2013_Biochem.J_454_387 |
| Author(s) : Masson P , Lushchekina SV , Schopfer LM , Lockridge O |
| Ref : Biochemical Journal , 454 :387 , 2013 |
| Abstract : Masson_2013_Biochem.J_454_387 |
| ESTHER : Masson_2013_Biochem.J_454_387 |
| PubMedSearch : Masson_2013_Biochem.J_454_387 |
| PubMedID: 23782236 |
| Title : Identifying safer anti-wear triaryl phosphate additives for jet engine lubricants - Baker_2013_Chem.Biol.Interact_203_257 |
| Author(s) : Baker PE , Cole TB , Cartwright M , Suzuki SM , Thummel KE , Lin YS , Co AL , Rettie AE , Kim JH , Furlong CE |
| Ref : Chemico-Biological Interactions , 203 :257 , 2013 |
| Abstract : Baker_2013_Chem.Biol.Interact_203_257 |
| ESTHER : Baker_2013_Chem.Biol.Interact_203_257 |
| PubMedSearch : Baker_2013_Chem.Biol.Interact_203_257 |
| PubMedID: 23085349 |
| Title : Cresyl saligenin phosphate, an organophosphorus toxicant, makes covalent adducts with histidine, lysine, and tyrosine residues of human serum albumin - Liyasova_2012_Chem.Res.Toxicol_25_1752 |
| Author(s) : Liyasova MS , Schopfer LM , Lockridge O |
| Ref : Chemical Research in Toxicology , 25 :1752 , 2012 |
| Abstract : Liyasova_2012_Chem.Res.Toxicol_25_1752 |
| ESTHER : Liyasova_2012_Chem.Res.Toxicol_25_1752 |
| PubMedSearch : Liyasova_2012_Chem.Res.Toxicol_25_1752 |
| PubMedID: 22793878 |
| Title : Reaction of cresyl saligenin phosphate, the organophosphorus agent implicated in aerotoxic syndrome, with human cholinesterases: mechanistic studies employing kinetics, mass spectrometry, and X-ray structure analysis - Carletti_2011_Chem.Res.Toxicol_24_797 |
| Author(s) : Carletti E , Schopfer LM , Colletier JP , Froment MT , Nachon F , Weik M , Lockridge O , Masson P |
| Ref : Chemical Research in Toxicology , 24 :797 , 2011 |
| Abstract : Carletti_2011_Chem.Res.Toxicol_24_797 |
| ESTHER : Carletti_2011_Chem.Res.Toxicol_24_797 |
| PubMedSearch : Carletti_2011_Chem.Res.Toxicol_24_797 |
| PubMedID: 21438623 |
| Gene_locus related to this paper: human-BCHE |
| Title : A murine model for sarin exposure using the carboxylesterase inhibitor CBDP - Garrett_2010_Neurotoxicol_31_502 |
| Author(s) : Garrett TL , Rapp CM , Grubbs RD , Schlager JJ , Lucot JB |
| Ref : Neurotoxicology , 31 :502 , 2010 |
| Abstract : Garrett_2010_Neurotoxicol_31_502 |
| ESTHER : Garrett_2010_Neurotoxicol_31_502 |
| PubMedSearch : Garrett_2010_Neurotoxicol_31_502 |
| PubMedID: 20510297 |
| Title : Development of diagnostics in the search for an explanation of aerotoxic syndrome - Schopfer_2010_Anal.Biochem_404_64 |
| Author(s) : Schopfer LM , Furlong CE , Lockridge O |
| Ref : Analytical Biochemistry , 404 :64 , 2010 |
| Abstract : Schopfer_2010_Anal.Biochem_404_64 |
| ESTHER : Schopfer_2010_Anal.Biochem_404_64 |
| PubMedSearch : Schopfer_2010_Anal.Biochem_404_64 |
| PubMedID: 20447373 |
| Title : Comparison of polyethylene glycol-conjugated recombinant human acetylcholinesterase and serum human butyrylcholinesterase as bioscavengers of organophosphate compounds - Cohen_2006_Mol.Pharmacol_70_1121 |
| Author(s) : Cohen O , Kronman C , Raveh L , Mazor O , Ordentlich A , Shafferman A |
| Ref : Molecular Pharmacology , 70 :1121 , 2006 |
| Abstract : Cohen_2006_Mol.Pharmacol_70_1121 |
| ESTHER : Cohen_2006_Mol.Pharmacol_70_1121 |
| PubMedSearch : Cohen_2006_Mol.Pharmacol_70_1121 |
| PubMedID: 16801396 |
| Title : Mechanism of differential inhibition of hepatic and pancreatic fatty acid ethyl ester synthase by inhibitors of serine-esterases: in vitro and cell culture studies - Kaphalia_2004_Toxicol.Appl.Pharmacol_200_7 |
| Author(s) : Kaphalia BS , Mericle KA , Ansari GA |
| Ref : Toxicol Appl Pharmacol , 200 :7 , 2004 |
| Abstract : Kaphalia_2004_Toxicol.Appl.Pharmacol_200_7 |
| ESTHER : Kaphalia_2004_Toxicol.Appl.Pharmacol_200_7 |
| PubMedSearch : Kaphalia_2004_Toxicol.Appl.Pharmacol_200_7 |
| PubMedID: 15451303 |
| Title : Different role of carboxylesterases in toxicity and tolerance to paraoxon and DFP - Dettbarn_1999_Chem.Biol.Interact_119-120_445 |
| Author(s) : Dettbarn WD , Yang ZP , Milatovic D |
| Ref : Chemico-Biological Interactions , 119-120 :445 , 1999 |
| Abstract : Dettbarn_1999_Chem.Biol.Interact_119-120_445 |
| ESTHER : Dettbarn_1999_Chem.Biol.Interact_119-120_445 |
| PubMedSearch : Dettbarn_1999_Chem.Biol.Interact_119-120_445 |
| PubMedID: 10421482 |
| Title : Prevention of tolerance to the organophosphorus anticholinesterase paraoxon with carboxylesterase inhibitors - Yang_1998_Biochem.Pharmacol_55_1419 |
| Author(s) : Yang ZP , Dettbarn WD |
| Ref : Biochemical Pharmacology , 55 :1419 , 1998 |
| Abstract : Yang_1998_Biochem.Pharmacol_55_1419 |
| ESTHER : Yang_1998_Biochem.Pharmacol_55_1419 |
| PubMedSearch : Yang_1998_Biochem.Pharmacol_55_1419 |
| PubMedID: 10076534 |
| Title : The in vitro degradation of cisatracurium, the R, cis-R'-isomer of atracurium, in human and rat plasma - Welch_1995_Clin.Pharmacol.Ther_58_132 |
| Author(s) : Welch RM , Brown A , Ravitch J , Dahl R |
| Ref : Clinical Pharmacology & Therapeutics , 58 :132 , 1995 |
| Abstract : Welch_1995_Clin.Pharmacol.Ther_58_132 |
| ESTHER : Welch_1995_Clin.Pharmacol.Ther_58_132 |
| PubMedSearch : Welch_1995_Clin.Pharmacol.Ther_58_132 |
| PubMedID: 7648763 |
| Title : Neuropathy target esterase inhibitors: enantiomeric separation and stereospecificity of 2-substituted-4H-1,3,2-benzodioxaphosphorin 2-oxides - Wu_1994_Chem.Res.Toxicol_7_77 |
| Author(s) : Wu SY , Casida JE |
| Ref : Chemical Research in Toxicology , 7 :77 , 1994 |
| Abstract : Wu_1994_Chem.Res.Toxicol_7_77 |
| ESTHER : Wu_1994_Chem.Res.Toxicol_7_77 |
| PubMedSearch : Wu_1994_Chem.Res.Toxicol_7_77 |
| PubMedID: 8155829 |
| Title : A purified recombinant organophosphorus acid anhydrase protects mice against soman - Broomfield_1993_Chem.Biol.Interact_87_279 |
| Author(s) : Broomfield CA |
| Ref : Chemico-Biological Interactions , 87 :279 , 1993 |
| Abstract : Broomfield_1993_Chem.Biol.Interact_87_279 |
| ESTHER : Broomfield_1993_Chem.Biol.Interact_87_279 |
| PubMedSearch : Broomfield_1993_Chem.Biol.Interact_87_279 |
| PubMedID: 8393744 |
| Title : Effect of pretreatment with CBDP on the toxicokinetics of soman stereoisomers in rats and guinea pigs - Due_1993_Arch.Toxicol_67_706 |
| Author(s) : Due AH , Trap HC , van der Wiel HJ , Benschop HP |
| Ref : Archives of Toxicology , 67 :706 , 1993 |
| Abstract : Due_1993_Arch.Toxicol_67_706 |
| ESTHER : Due_1993_Arch.Toxicol_67_706 |
| PubMedSearch : Due_1993_Arch.Toxicol_67_706 |
| PubMedID: 8135661 |
| Title : Toxicokinetics of [14C]-saligenin cyclic-o-tolyl phosphate in anesthetized male F-344 rats - Burka_1993_Reprod.Toxicol_7_81 |
| Author(s) : Burka LT , Chapin RE |
| Ref : Reprod Toxicol , 7 :81 , 1993 |
| Abstract : Burka_1993_Reprod.Toxicol_7_81 |
| ESTHER : Burka_1993_Reprod.Toxicol_7_81 |
| PubMedSearch : Burka_1993_Reprod.Toxicol_7_81 |
| PubMedID: 8448420 |
| Title : Local application of neuropathic organophosphorus compounds to hen sciatic nerve: inhibition of neuropathy target esterase and peripheral neurological impairments - Carrera_1992_Toxicol.Appl.Pharmacol_117_218 |
| Author(s) : Carrera V , Barril J , Mauricio M , Pellin M , Vilanova E |
| Ref : Toxicol Appl Pharmacol , 117 :218 , 1992 |
| Abstract : Carrera_1992_Toxicol.Appl.Pharmacol_117_218 |
| ESTHER : Carrera_1992_Toxicol.Appl.Pharmacol_117_218 |
| PubMedSearch : Carrera_1992_Toxicol.Appl.Pharmacol_117_218 |
| PubMedID: 1471154 |
| Title : Murine susceptibility to organophosphorus-induced delayed neuropathy (OPIDN) - Veronesi_1991_Toxicol.Appl.Pharmacol_107_311 |
| Author(s) : Veronesi B , Padilla S , Blackmon K , Pope C |
| Ref : Toxicol Appl Pharmacol , 107 :311 , 1991 |
| Abstract : Veronesi_1991_Toxicol.Appl.Pharmacol_107_311 |
| ESTHER : Veronesi_1991_Toxicol.Appl.Pharmacol_107_311 |
| PubMedSearch : Veronesi_1991_Toxicol.Appl.Pharmacol_107_311 |
| PubMedID: 1994513 |
| Title : The effects of tri-o-cresyl phosphate and metabolites on rat Sertoli cell function in primary culture - Chapin_1991_Toxicol.Appl.Pharmacol_108_194 |
| Author(s) : Chapin RE , Phelps JL , Burka LT , Abou-Donia MB , Heindel JJ |
| Ref : Toxicol Appl Pharmacol , 108 :194 , 1991 |
| Abstract : Chapin_1991_Toxicol.Appl.Pharmacol_108_194 |
| ESTHER : Chapin_1991_Toxicol.Appl.Pharmacol_108_194 |
| PubMedSearch : Chapin_1991_Toxicol.Appl.Pharmacol_108_194 |
| PubMedID: 1902005 |
| Title : Serum carboxylesterase activity in various strains of rats: sensitivity to inhibition by CBDP (2-\/o-cresyl\/4H:1:3:2-benzodioxaphosphorin-2-oxide) - Clement_1990_Arch.Toxicol_64_414 |
| Author(s) : Clement JG , Erhardt N |
| Ref : Archives of Toxicology , 64 :414 , 1990 |
| Abstract : Clement_1990_Arch.Toxicol_64_414 |
| ESTHER : Clement_1990_Arch.Toxicol_64_414 |
| PubMedSearch : Clement_1990_Arch.Toxicol_64_414 |
| PubMedID: 2403290 |
| Title : Effects of CBDP and MEPQ on the toxicity and distribution of [3H]-soman in mice - Shapira_1990_Arch.Toxicol_64_663 |
| Author(s) : Shapira S , Kadar T , Cohen G , Chapman S , Raveh L |
| Ref : Archives of Toxicology , 64 :663 , 1990 |
| Abstract : Shapira_1990_Arch.Toxicol_64_663 |
| ESTHER : Shapira_1990_Arch.Toxicol_64_663 |
| PubMedSearch : Shapira_1990_Arch.Toxicol_64_663 |
| PubMedID: 2090035 |
| Title : The interaction of Sertoli and Leydig cells in the testicular toxicity of tri-o-cresyl phosphate - Chapin_1990_Toxicol.Appl.Pharmacol_104_483 |
| Author(s) : Chapin RE , Phelps JL , Somkuti SG , Heindel JJ , Burka LT |
| Ref : Toxicol Appl Pharmacol , 104 :483 , 1990 |
| Abstract : Chapin_1990_Toxicol.Appl.Pharmacol_104_483 |
| ESTHER : Chapin_1990_Toxicol.Appl.Pharmacol_104_483 |
| PubMedSearch : Chapin_1990_Toxicol.Appl.Pharmacol_104_483 |
| PubMedID: 2385838 |
| Title : Hepatic subcellular localization of cresylbenzodioxaphosphorin oxide (CBDP)-sensitive soman binding sites - Little_1990_Biochem.Pharmacol_40_1733 |
| Author(s) : Little JS , Maxwell DM , Fox-Talbot MK , Brecht K , Lenz DE |
| Ref : Biochemical Pharmacology , 40 :1733 , 1990 |
| Abstract : Little_1990_Biochem.Pharmacol_40_1733 |
| ESTHER : Little_1990_Biochem.Pharmacol_40_1733 |
| PubMedSearch : Little_1990_Biochem.Pharmacol_40_1733 |
| PubMedID: 2242010 |
| Title : The effect of 2-(o-cresyl)-4H-1:3:2-benzodioxaphosphorin-2-oxide on tissue cholinesterase and carboxylesterase activities of the rat - Jimmerson_1989_Fundam.Appl.Toxicol_13_568 |
| Author(s) : Jimmerson VR , Shih TM , Maxwell DM , Kaminskis A , Mailman RB |
| Ref : Fundamental & Applied Toxicology , 13 :568 , 1989 |
| Abstract : Jimmerson_1989_Fundam.Appl.Toxicol_13_568 |
| ESTHER : Jimmerson_1989_Fundam.Appl.Toxicol_13_568 |
| PubMedSearch : Jimmerson_1989_Fundam.Appl.Toxicol_13_568 |
| PubMedID: 2612789 |
| Title : Cresylbenzodioxaphosphorin oxide pretreatment alters soman-induced toxicity and inhibition of tissue cholinesterase activity of the rat - Jimmerson_1989_Toxicol.Lett_48_93 |
| Author(s) : Jimmerson VR , Shih TM , Maxwell DM , Mailman RB |
| Ref : Toxicol Lett , 48 :93 , 1989 |
| Abstract : Jimmerson_1989_Toxicol.Lett_48_93 |
| ESTHER : Jimmerson_1989_Toxicol.Lett_48_93 |
| PubMedSearch : Jimmerson_1989_Toxicol.Lett_48_93 |
| PubMedID: 2749782 |
| Title : Effect of beta-naphthoflavone on o-tolyl saligenin phosphate-induced delayed neuropathy in two lines of chickens - Bursian_1989_J.Toxicol.Environ.Health_28_461 |
| Author(s) : Bursian SJ , Lehning EJ , Correll L , Ehrich M |
| Ref : J Toxicol Environ Health , 28 :461 , 1989 |
| Abstract : Bursian_1989_J.Toxicol.Environ.Health_28_461 |
| ESTHER : Bursian_1989_J.Toxicol.Environ.Health_28_461 |
| PubMedSearch : Bursian_1989_J.Toxicol.Environ.Health_28_461 |
| PubMedID: 2593176 |
| Title : Effect of carboxylesterase inhibition on carbamate protection against soman toxicity - Maxwell_1988_J.Pharmacol.Exp.Ther_246_986 |
| Author(s) : Maxwell DM , Brecht KM , Lenz DE , O'Neill BL |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 246 :986 , 1988 |
| Abstract : Maxwell_1988_J.Pharmacol.Exp.Ther_246_986 |
| ESTHER : Maxwell_1988_J.Pharmacol.Exp.Ther_246_986 |
| PubMedSearch : Maxwell_1988_J.Pharmacol.Exp.Ther_246_986 |
| PubMedID: 3418520 |
| Title : Effect of route of administration on the development of organophosphate-induced delayed neurotoxicity in 4-week-old chicks - Olson_1988_J.Toxicol.Environ.Health_23_499 |
| Author(s) : Olson BA , Bursian SJ |
| Ref : J Toxicol Environ Health , 23 :499 , 1988 |
| Abstract : Olson_1988_J.Toxicol.Environ.Health_23_499 |
| ESTHER : Olson_1988_J.Toxicol.Environ.Health_23_499 |
| PubMedSearch : Olson_1988_J.Toxicol.Environ.Health_23_499 |
| PubMedID: 3361617 |
| Title : Stereoisomers of soman (pinacolyl methylphosphonofluoridate): inhibition of serum carboxylic ester hydrolase and potentiation of their toxicity by CBDP (2-(2-methylphenoxy)-4H-1,3,2-benzodioxaphosphorin-2-oxide) in mice - Clement_1987_Toxicol.Appl.Pharmacol_89_141 |
| Author(s) : Clement JG , Benschop HP , DeJong LP , Wolthuis OL |
| Ref : Toxicol Appl Pharmacol , 89 :141 , 1987 |
| Abstract : Clement_1987_Toxicol.Appl.Pharmacol_89_141 |
| ESTHER : Clement_1987_Toxicol.Appl.Pharmacol_89_141 |
| PubMedSearch : Clement_1987_Toxicol.Appl.Pharmacol_89_141 |
| PubMedID: 3590185 |
| Title : The effect of carboxylesterase inhibition on interspecies differences in soman toxicity - Maxwell_1987_Toxicol.Lett_39_35 |
| Author(s) : Maxwell DM , Brecht KM , O'Neill BL |
| Ref : Toxicol Lett , 39 :35 , 1987 |
| Abstract : Maxwell_1987_Toxicol.Lett_39_35 |
| ESTHER : Maxwell_1987_Toxicol.Lett_39_35 |
| PubMedSearch : Maxwell_1987_Toxicol.Lett_39_35 |
| PubMedID: 3672554 |
| Title : Role of aliesterase in organophosphate poisoning - Clement_1984_Fundam.Appl.Toxicol_4_S96 |
| Author(s) : Clement JG |
| Ref : Fundamental & Applied Toxicology , 4 :S96 , 1984 |
| Abstract : Clement_1984_Fundam.Appl.Toxicol_4_S96 |
| ESTHER : Clement_1984_Fundam.Appl.Toxicol_4_S96 |
| PubMedSearch : Clement_1984_Fundam.Appl.Toxicol_4_S96 |
| PubMedID: 6724216 |
| Title : Importance of aliesterase as a detoxification mechanism for soman (Pinacolyl methylphosphonofluoridate) in mice - Clement_1984_Biochem.Pharmacol_33_3807 |
| Author(s) : Clement JG |
| Ref : Biochemical Pharmacology , 33 :3807 , 1984 |
| Abstract : Clement_1984_Biochem.Pharmacol_33_3807 |
| ESTHER : Clement_1984_Biochem.Pharmacol_33_3807 |
| PubMedSearch : Clement_1984_Biochem.Pharmacol_33_3807 |
| PubMedID: 6508835 |
| Title : Antidote effect of sodium fluoride against organophosphate poisoning in mice - Clement_1983_Life.Sci_32_1803 |
| Author(s) : Clement JG , Filbert M , Filbert MG |
| Ref : Life Sciences , 32 :1803 , 1983 |
| Abstract : Clement_1983_Life.Sci_32_1803 |
| ESTHER : Clement_1983_Life.Sci_32_1803 |
| PubMedSearch : Clement_1983_Life.Sci_32_1803 |
| PubMedID: 6835011 |
| Title : The influence of 2-\/o-cresyl\/-4 H-1 : 3 : 2-benzodioxa-phosphorin-2-oxide (CBDP) on organophosphate poisoning and its therapy - Boskovic_1979_Arch.Toxicol_42_207 |
| Author(s) : Boskovic B |
| Ref : Archives of Toxicology , 42 :207 , 1979 |
| Abstract : Boskovic_1979_Arch.Toxicol_42_207 |
| ESTHER : Boskovic_1979_Arch.Toxicol_42_207 |
| PubMedSearch : Boskovic_1979_Arch.Toxicol_42_207 |
| PubMedID: 475595 |
| Title : Biological activity of a tri-o-cresyl phosphate metabolite - Casida_1961_Nature_191_1396 |
| Author(s) : Casida JE , Eto M , Baron RL |
| Ref : Nature , 191 :1396 , 1961 |
| Abstract : Casida_1961_Nature_191_1396 |
| ESTHER : Casida_1961_Nature_191_1396 |
| PubMedSearch : Casida_1961_Nature_191_1396 |
| PubMedID: |