Galanthamine
In 1951, the Russian pharmacologist Mikhail Mashkovsky discovered that local villagers from the Urals used the wild snowdrop (Galanthus spp., most notably Galanthus woronowii Losinsk.) to stave off paralysis in children suffering from poliomyelitis. The ethnobotanical use of the snowdrop in the Urals led to the isolation of galanthamine for the first time in 1952, an alkaloid with AChE-inhibiting propertiesAlkaloid from caucasian snow-drop Galenthus woronowii, the common snowdrop Galanthus nivalis Ungernia genus, Amaryllidaceae, from Narcissus spp. Selectivity for AChE. The CAS number of Galanthamine Hydrobromide is 1953-04-4. || Nivalin Reminyl trademark approved for treatment of Alzheimer disease in Austria. || Galantamine is a specific competitive and reversible aceylcholinesterase inhibitor. It is also an allosteric modulator at nicotinic cholinergic receptor sites potentiating cholinergic nicotinic neurotransmission at the ganglionic and the neuromuscular junctions.
General
Type : Derivative of Galanthamine,Natural,Alkaloid,Drug,Benzazepin,Azepine
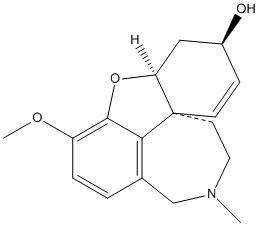
Chemical_Nomenclature : 1,2,3,4,6,7,7a,11c-octahydro-9-methoxy-2methylbenzofuro[4,3,2-efg][2]benzazocin-6-ol
Canonical SMILES : CN1CCC23C=CC(CC2OC4=C(C=CC(=C34)C1)OC)O
InChI : InChI=1S\/C17H21NO3\/c1-18-8-7-17-6-5-12(19)9-14(17)21-16-13(20-2)4-3-11(10-18)15(16)17\/h3-6,12,14,19H,7-10H2,1-2H3\/t12-,14-,17-\/m0\/s1
InChIKey : ASUTZQLVASHGKV-JDFRZJQESA-N
Other name(s) : Galantamine,Lycoremine,Jilkon,Nivalin,Reminyl,Razadyne
MW : 287.35
Formula : C17H21NO3
CAS_number : 357-70-0
PubChem : 9651
UniChem : ASUTZQLVASHGKV-JDFRZJQESA-N
IUPHAR : 6693
Wikipedia : Galanthamine
Target
Families : Galanthamine ligand of proteins in family: ACHE
Stucture :
Protein : torca-ACHE || human-ACHE
References (45)
| Title : Galantamine improves glycemic control and diabetic nephropathy in Lepr(db\/db) mice - Meng_2023_Sci.Rep_13_15544 |
| Author(s) : Meng Q , Ma J , Suo L , Pruekprasert N , Chakrapani P , Cooney RN |
| Ref : Sci Rep , 13 :15544 , 2023 |
| Abstract : Meng_2023_Sci.Rep_13_15544 |
| ESTHER : Meng_2023_Sci.Rep_13_15544 |
| PubMedSearch : Meng_2023_Sci.Rep_13_15544 |
| PubMedID: 37731032 |
| Title : Chemical Survey of Three Species of the Genus Rauhia Traub (Amaryllidaceae) - Tallini_2022_Plants.(Basel)_11_ |
| Author(s) : Tallini LR , Osorio EH , Berkov S , Torras-Claveria L , Rodriguez-Escobar ML , Viladomat F , Meerow AW , Bastida J |
| Ref : Plants (Basel) , 11 : , 2022 |
| Abstract : Tallini_2022_Plants.(Basel)_11_ |
| ESTHER : Tallini_2022_Plants.(Basel)_11_ |
| PubMedSearch : Tallini_2022_Plants.(Basel)_11_ |
| PubMedID: 36559661 |
| Title : Loss of Basal Forebrain Cholinergic Neurons Following Adolescent Binge Ethanol Exposure: Recovery With the Cholinesterase Inhibitor Galantamine - Crews_2021_Front.Behav.Neurosci_15_652494 |
| Author(s) : Crews FT , Fisher R , Deason C , Vetreno RP |
| Ref : Front Behavioral Neuroscience , 15 :652494 , 2021 |
| Abstract : Crews_2021_Front.Behav.Neurosci_15_652494 |
| ESTHER : Crews_2021_Front.Behav.Neurosci_15_652494 |
| PubMedSearch : Crews_2021_Front.Behav.Neurosci_15_652494 |
| PubMedID: 33716687 |
| Title : Gigantelline, gigantellinine and gigancrinine, cherylline- and crinine-type alkaloids isolated from Crinum jagus with anti-acetylcholinesterase activity - Ka_2020_Phytochemistry_175_112390 |
| Author(s) : Ka S , Masi M , Merindol N , Di Lecce R , Plourde MB , Seck M , Gorecki M , Pescitelli G , Desgagne-Penix I , Evidente A |
| Ref : Phytochemistry , 175 :112390 , 2020 |
| Abstract : Ka_2020_Phytochemistry_175_112390 |
| ESTHER : Ka_2020_Phytochemistry_175_112390 |
| PubMedSearch : Ka_2020_Phytochemistry_175_112390 |
| PubMedID: 32335411 |
| Title : Cholinesterase-inhibitory effect and in silico analysis of alkaloids from bulbs of Hieronymiella species - Ortiz_2018_Phytomedicine_39_66 |
| Author(s) : Ortiz JE , Garro A , Pigni NB , Aguero MB , Roitman G , Slanis A , Enriz RD , Feresin GE , Bastida J , Tapia A |
| Ref : Phytomedicine , 39 :66 , 2018 |
| Abstract : Ortiz_2018_Phytomedicine_39_66 |
| ESTHER : Ortiz_2018_Phytomedicine_39_66 |
| PubMedSearch : Ortiz_2018_Phytomedicine_39_66 |
| PubMedID: 29433685 |
| Title : Cooperative hydrogen bonds and mobility of the non-aromatic ring as selectivity determinants for human acetylcholinesterase to similar anti-Alzheimer's galantaminics: a computational study - Rocha_2018_J.Biomol.Struct.Dyn__1 |
| Author(s) : Rocha REO , Lima LHF |
| Ref : J Biomol Struct Dyn , :1 , 2018 |
| Abstract : Rocha_2018_J.Biomol.Struct.Dyn__1 |
| ESTHER : Rocha_2018_J.Biomol.Struct.Dyn__1 |
| PubMedSearch : Rocha_2018_J.Biomol.Struct.Dyn__1 |
| PubMedID: 29697300 |
| Title : Post-Injury Administration of Galantamine Reduces Traumatic Brain Injury Pathology and Improves Outcome - Zhao_2018_J.Neurotrauma_35_362 |
| Author(s) : Zhao J , Hylin MJ , Kobori N , Hood KN , Moore AN , Dash PK |
| Ref : Journal of Neurotrauma , 35 :362 , 2018 |
| Abstract : Zhao_2018_J.Neurotrauma_35_362 |
| ESTHER : Zhao_2018_J.Neurotrauma_35_362 |
| PubMedSearch : Zhao_2018_J.Neurotrauma_35_362 |
| PubMedID: 29088998 |
| Title : Alkaloids from Habranthus tubispathus and H. jamesonii, two amaryllidaceae with acetyl- and butyrylcholinesterase inhibition activity - Cavallaro_2014_Nat.Prod.Commun_9_159 |
| Author(s) : Cavallaro V , Alza NP , Murray MG , Murray AP |
| Ref : Nat Prod Commun , 9 :159 , 2014 |
| Abstract : Cavallaro_2014_Nat.Prod.Commun_9_159 |
| ESTHER : Cavallaro_2014_Nat.Prod.Commun_9_159 |
| PubMedSearch : Cavallaro_2014_Nat.Prod.Commun_9_159 |
| PubMedID: 24689279 |
| Title : Galantamine potentiates the neuroprotective effect of memantine against NMDA-induced excitotoxicity - Lopes_2013_Brain.Behav_3_67 |
| Author(s) : Lopes JP , Tarozzo G , Reggiani A , Piomelli D , Cavalli A |
| Ref : Brain Behav , 3 :67 , 2013 |
| Abstract : Lopes_2013_Brain.Behav_3_67 |
| ESTHER : Lopes_2013_Brain.Behav_3_67 |
| PubMedSearch : Lopes_2013_Brain.Behav_3_67 |
| PubMedID: 23532860 |
| Title : Structures of human acetylcholinesterase in complex with pharmacologically important ligands - Cheung_2012_J.Med.Chem_55_10282 |
| Author(s) : Cheung J , Rudolph MJ , Burshteyn F , Cassidy MS , Gary EN , Love J , Franklin MC , Height JJ |
| Ref : Journal of Medicinal Chemistry , 55 :10282 , 2012 |
| Abstract : Cheung_2012_J.Med.Chem_55_10282 |
| ESTHER : Cheung_2012_J.Med.Chem_55_10282 |
| PubMedSearch : Cheung_2012_J.Med.Chem_55_10282 |
| PubMedID: 23035744 |
| Gene_locus related to this paper: human-ACHE |
| Title : GC-MS of amaryllidaceous galanthamine-type alkaloids - Berkov_2012_J.Mass.Spectrom_47_1065 |
| Author(s) : Berkov S , Viladomat F , Codina C , Suarez S , Ravelo A , Bastida J |
| Ref : J Mass Spectrom , 47 :1065 , 2012 |
| Abstract : Berkov_2012_J.Mass.Spectrom_47_1065 |
| ESTHER : Berkov_2012_J.Mass.Spectrom_47_1065 |
| PubMedSearch : Berkov_2012_J.Mass.Spectrom_47_1065 |
| PubMedID: 22899516 |
| Title : Rapid TLC\/GC-MS identification of acetylcholinesterase inhibitors in alkaloid extracts - Berkov_2008_Phytochem.Anal_19_411 |
| Author(s) : Berkov S , Bastida J , Nikolova M , Viladomat F , Codina C |
| Ref : Phytochem Anal , 19 :411 , 2008 |
| Abstract : Berkov_2008_Phytochem.Anal_19_411 |
| ESTHER : Berkov_2008_Phytochem.Anal_19_411 |
| PubMedSearch : Berkov_2008_Phytochem.Anal_19_411 |
| PubMedID: 18446766 |
| Title : Galanthamine from snowdrop--the development of a modern drug against Alzheimer's disease from local Caucasian knowledge - Heinrich_2004_J.Ethnopharmacol_92_147 |
| Author(s) : Heinrich M , Lee Teoh H |
| Ref : J Ethnopharmacol , 92 :147 , 2004 |
| Abstract : Heinrich_2004_J.Ethnopharmacol_92_147 |
| ESTHER : Heinrich_2004_J.Ethnopharmacol_92_147 |
| PubMedSearch : Heinrich_2004_J.Ethnopharmacol_92_147 |
| PubMedID: 15137996 |
| Title : The complex of a bivalent derivative of galanthamine with torpedo acetylcholinesterase displays drastic deformation of the active-site gorge: implications for structure-based drug design - Greenblatt_2004_J.Am.Chem.Soc_126_15405 |
| Author(s) : Greenblatt HM , Guillou C , Guenard D , Argaman A , Botti SA , Badet B , Thal C , Silman I , Sussman JL |
| Ref : Journal of the American Chemical Society , 126 :15405 , 2004 |
| Abstract : Greenblatt_2004_J.Am.Chem.Soc_126_15405 |
| ESTHER : Greenblatt_2004_J.Am.Chem.Soc_126_15405 |
| PubMedSearch : Greenblatt_2004_J.Am.Chem.Soc_126_15405 |
| PubMedID: 15563167 |
| Gene_locus related to this paper: torca-ACHE |
| Title : Acetylcholinesterase inhibitory activity of some Amaryllidaceae alkaloids and Narcissus extracts - Lopez_2002_Life.Sci_71_2521 |
| Author(s) : Lopez S , Bastida J , Viladomat F , Codina C |
| Ref : Life Sciences , 71 :2521 , 2002 |
| Abstract : Lopez_2002_Life.Sci_71_2521 |
| ESTHER : Lopez_2002_Life.Sci_71_2521 |
| PubMedSearch : Lopez_2002_Life.Sci_71_2521 |
| PubMedID: 12270757 |
| Title : Three-dimensional structure of a complex of galanthamine (Nivalin) with acetylcholinesterase from Torpedo californica: implications for the design of new anti-Alzheimer drugs - Bartolucci_2001_Proteins_42_182 |
| Author(s) : Bartolucci C , Perola E , Pilger C , Fels G , Lamba D |
| Ref : Proteins , 42 :182 , 2001 |
| Abstract : Bartolucci_2001_Proteins_42_182 |
| ESTHER : Bartolucci_2001_Proteins_42_182 |
| PubMedSearch : Bartolucci_2001_Proteins_42_182 |
| PubMedID: 11119642 |
| Gene_locus related to this paper: torca-ACHE |
| Title : Potent acetylcholinesterase inhibitors: design, synthesis and structure-activity relationships of alkylene linked bis-galanthamine and galanthamine-galanthaminium salts - Guillou_2000_Bioorg.Med.Chem.Lett_10_637 |
| Author(s) : Guillou C , Mary A , Renko DZ , Gras E , Thal C |
| Ref : Bioorganic & Medicinal Chemistry Lett , 10 :637 , 2000 |
| Abstract : Guillou_2000_Bioorg.Med.Chem.Lett_10_637 |
| ESTHER : Guillou_2000_Bioorg.Med.Chem.Lett_10_637 |
| PubMedSearch : Guillou_2000_Bioorg.Med.Chem.Lett_10_637 |
| PubMedID: 10762042 |
| Title : Effect of the acetylcholinesterase inhibitor galanthamine on learning and memory in prolonged alcohol intake rat model of acetylcholine deficit - Iliev_1999_Methods.Find.Exp.Clin.Pharmacol_21_297 |
| Author(s) : Iliev A , Traykov V , Prodanov D , Mantchev G , Yakimova K , Krushkov I , Boyadjieva N |
| Ref : Methods Find Exp Clin Pharmacol , 21 :297 , 1999 |
| Abstract : Iliev_1999_Methods.Find.Exp.Clin.Pharmacol_21_297 |
| ESTHER : Iliev_1999_Methods.Find.Exp.Clin.Pharmacol_21_297 |
| PubMedSearch : Iliev_1999_Methods.Find.Exp.Clin.Pharmacol_21_297 |
| PubMedID: 10399139 |
| Title : Capillary zone electrophoresis determination of galanthamine in biological fluids and pharmaceutical preparatives: experimental design and artificial neural network optimization - Pokorna_1999_Electrophoresis_20_1993 |
| Author(s) : Pokorna L , Revilla A , Havel J , Patocka J |
| Ref : Electrophoresis , 20 :1993 , 1999 |
| Abstract : Pokorna_1999_Electrophoresis_20_1993 |
| ESTHER : Pokorna_1999_Electrophoresis_20_1993 |
| PubMedSearch : Pokorna_1999_Electrophoresis_20_1993 |
| PubMedID: 10451107 |
| Title : The O-demethylation of the antidementia drug galanthamine is catalysed by cytochrome P450 2D6 - Bachus_1999_Pharmacogenet_9_661 |
| Author(s) : Bachus R , Bickel U , Thomsen T , Roots I , Kewitz H |
| Ref : Pharmacogenetics , 9 :661 , 1999 |
| Abstract : Bachus_1999_Pharmacogenet_9_661 |
| ESTHER : Bachus_1999_Pharmacogenet_9_661 |
| PubMedSearch : Bachus_1999_Pharmacogenet_9_661 |
| PubMedID: 10634129 |
| Title : Alzheimer's disease: seeking new ways to preserve brain function. Interview by Alice V. Luddington - Davis_1999_Geriatrics_54_42 |
| Author(s) : Davis KL |
| Ref : Geriatrics , 54 :42 , 1999 |
| Abstract : Davis_1999_Geriatrics_54_42 |
| ESTHER : Davis_1999_Geriatrics_54_42 |
| PubMedSearch : Davis_1999_Geriatrics_54_42 |
| PubMedID: 10024872 |
| Title : Structure of acetylcholinesterase complexed with (-)-galanthamine at 2.3 A resolution - Greenblatt_1999_FEBS.Lett_463_321 |
| Author(s) : Greenblatt HM , Kryger G , Lewis T , Silman I , Sussman JL |
| Ref : FEBS Letters , 463 :321 , 1999 |
| Abstract : Greenblatt_1999_FEBS.Lett_463_321 |
| ESTHER : Greenblatt_1999_FEBS.Lett_463_321 |
| PubMedSearch : Greenblatt_1999_FEBS.Lett_463_321 |
| PubMedID: 10606746 |
| Gene_locus related to this paper: torca-ACHE |
| Title : Potent acetylcholinesterase inhibitors: design, synthesis, and structure-activity relationships of bis-interacting ligands in the galanthamine series - Mary_1998_Bioorg.Med.Chem_6_1835 |
| Author(s) : Mary A , Renko DZ , Guillou C , Thal C |
| Ref : Bioorganic & Medicinal Chemistry , 6 :1835 , 1998 |
| Abstract : Mary_1998_Bioorg.Med.Chem_6_1835 |
| ESTHER : Mary_1998_Bioorg.Med.Chem_6_1835 |
| PubMedSearch : Mary_1998_Bioorg.Med.Chem_6_1835 |
| PubMedID: 9839013 |
| Title : Invited review: Cholinesterase inhibitors for Alzheimer's disease therapy: from tacrine to future applications - Giacobini_1998_Neurochem.Int_32_413 |
| Author(s) : Giacobini E |
| Ref : Neurochem Int , 32 :413 , 1998 |
| Abstract : Giacobini_1998_Neurochem.Int_32_413 |
| ESTHER : Giacobini_1998_Neurochem.Int_32_413 |
| PubMedSearch : Giacobini_1998_Neurochem.Int_32_413 |
| PubMedID: 9676739 |
| Title : Evaluation of four Narcissus cultivars as potential sources for galanthamine production [letter] - Moraes-Cerdeira_1997_Planta.Med_63_472 |
| Author(s) : Moraes-Cerdeira RM , Burandt CL, Jr. , Bastos JK , Nanayakkara D , Mikell J , Thurn J , McChesney JD |
| Ref : Planta Med , 63 :472 , 1997 |
| Abstract : Moraes-Cerdeira_1997_Planta.Med_63_472 |
| ESTHER : Moraes-Cerdeira_1997_Planta.Med_63_472 |
| PubMedSearch : Moraes-Cerdeira_1997_Planta.Med_63_472 |
| PubMedID: 9342954 |
| Title : Pharmacological basis of drug therapy of Alzheimer's disease - Sharma_1997_Indian.J.Exp.Biol_35_1146 |
| Author(s) : Sharma A , Parikh V , Singh M |
| Ref : Indian J Exp Biol , 35 :1146 , 1997 |
| Abstract : Sharma_1997_Indian.J.Exp.Biol_35_1146 |
| ESTHER : Sharma_1997_Indian.J.Exp.Biol_35_1146 |
| PubMedSearch : Sharma_1997_Indian.J.Exp.Biol_35_1146 |
| PubMedID: 9567741 |
| Title : Pharmacological evaluation of novel Alzheimer's disease therapeutics: acetylcholinesterase inhibitors related to galanthamine - Bores_1996_J.Pharmacol.Exp.Ther_277_728 |
| Author(s) : Bores GM , Huger FP , Petko W , Mutlib AE , Camacho F , Rush DK , Selk DE , Wolf V , Kosley RW, Jr. , Davis L , Vargas HM |
| Ref : Journal of Pharmacology & Experimental Therapeutics , 277 :728 , 1996 |
| Abstract : Bores_1996_J.Pharmacol.Exp.Ther_277_728 |
| ESTHER : Bores_1996_J.Pharmacol.Exp.Ther_277_728 |
| PubMedSearch : Bores_1996_J.Pharmacol.Exp.Ther_277_728 |
| PubMedID: 8627552 |
| Title : Galanthamine. [Review] [23 refs] - Fulton_1996_Drugs.Aging_9_60 |
| Author(s) : Fulton B , Benfield P |
| Ref : Drugs & Aging , 9 :60 , 1996 |
| Abstract : Fulton_1996_Drugs.Aging_9_60 |
| ESTHER : Fulton_1996_Drugs.Aging_9_60 |
| PubMedSearch : Fulton_1996_Drugs.Aging_9_60 |
| PubMedID: 8818586 |
| Title : [The comparative clinico-experimental characteristics of aminostigmine and galanthamine used for treating poisonings by choline-blocking substances]. [Russian] - Prozorovskii_1996_Eksp.Klin.Farmakol_59_64 |
| Author(s) : Prozorovskii VB , Velikova VD , Pshenkina NN , Vasilenko ET |
| Ref : Eksperimentalnaia i Klinicheskaia Farmakologiia , 59 :64 , 1996 |
| Abstract : Prozorovskii_1996_Eksp.Klin.Farmakol_59_64 |
| ESTHER : Prozorovskii_1996_Eksp.Klin.Farmakol_59_64 |
| PubMedSearch : Prozorovskii_1996_Eksp.Klin.Farmakol_59_64 |
| PubMedID: 8704639 |
| Title : Effect of the cholinesterase-inhibiting substance galanthamine on evoked visual potentials in rats - Lewandowski_1995_Acta.Neurobiol.Experiment_55_141 |
| Author(s) : Lewandowski MH , Zmuda L |
| Ref : Acta Neurobiologiae Experimentalis , 55 :141 , 1995 |
| Abstract : Lewandowski_1995_Acta.Neurobiol.Experiment_55_141 |
| ESTHER : Lewandowski_1995_Acta.Neurobiol.Experiment_55_141 |
| PubMedSearch : Lewandowski_1995_Acta.Neurobiol.Experiment_55_141 |
| PubMedID: 7660864 |
| Title : Physostigmine, galanthamine and codeine act as 'noncompetitive nicotinic receptor agonists' on clonal rat pheochromocytoma cells - Storch_1995_Eur.J.Pharmacol_290_207 |
| Author(s) : Storch A , Schrattenholz A , Cooper JC , Abdel Ghani EM , Gutbrod O , Weber KH , Reinhardt S , Lobron C , Hermsen B , Soskic V , Pereira EF , Albuquerque EX , Methfessel C , Maelicke A |
| Ref : European Journal of Pharmacology , 290 :207 , 1995 |
| Abstract : Storch_1995_Eur.J.Pharmacol_290_207 |
| ESTHER : Storch_1995_Eur.J.Pharmacol_290_207 |
| PubMedSearch : Storch_1995_Eur.J.Pharmacol_290_207 |
| PubMedID: 7589215 |
| Title : The pharmacology of galanthamine and its analogues - Harvey_1995_Pharmacol.Ther_68_113 |
| Author(s) : Harvey AL |
| Ref : Pharmacol Ther , 68 :113 , 1995 |
| Abstract : Harvey_1995_Pharmacol.Ther_68_113 |
| ESTHER : Harvey_1995_Pharmacol.Ther_68_113 |
| PubMedSearch : Harvey_1995_Pharmacol.Ther_68_113 |
| PubMedID: 8604434 |
| Title : [Aminostigmine as a cholinesterase inhibitor and as an agent for treating poisonings by cholinergic blockers]. [Russian] - Prozorovskii_1994_Eksp.Klin.Farmakol_57_13 |
| Author(s) : Prozorovskii VB , Livanov GA , Velikova VD , Afanas'ev VV , Pavlova LV |
| Ref : Eksperimentalnaia i Klinicheskaia Farmakologiia , 57 :13 , 1994 |
| Abstract : Prozorovskii_1994_Eksp.Klin.Farmakol_57_13 |
| ESTHER : Prozorovskii_1994_Eksp.Klin.Farmakol_57_13 |
| PubMedSearch : Prozorovskii_1994_Eksp.Klin.Farmakol_57_13 |
| PubMedID: 7696893 |
| Title : Inhibition of acetylcholinesterase activity in human brain tissue and erythrocytes by galanthamine, physostigmine and tacrine - Thomsen_1991_Eur.J.Clin.Chem.Clin.Biochem_29_487 |
| Author(s) : Thomsen T , Kaden B , Fischer JP , Bickel U , Barz H , Gusztony G , Cervos-Navarro J , Kewitz H |
| Ref : European Journal of Clinical Chemistry & Clinical Biochemistry , 29 :487 , 1991 |
| Abstract : Thomsen_1991_Eur.J.Clin.Chem.Clin.Biochem_29_487 |
| ESTHER : Thomsen_1991_Eur.J.Clin.Chem.Clin.Biochem_29_487 |
| PubMedSearch : Thomsen_1991_Eur.J.Clin.Chem.Clin.Biochem_29_487 |
| PubMedID: 1954303 |
| Title : Selective inhibition of human acetylcholinesterase by galanthamine in vitro and in vivo - Thomsen_1990_Life.Sci_46_1553 |
| Author(s) : Thomsen T , Kewitz H |
| Ref : Life Sciences , 46 :1553 , 1990 |
| Abstract : Thomsen_1990_Life.Sci_46_1553 |
| ESTHER : Thomsen_1990_Life.Sci_46_1553 |
| PubMedSearch : Thomsen_1990_Life.Sci_46_1553 |
| PubMedID: 2355800 |
| Title : [Catalytic properties of cholinesterases immobilized in a gelatin membrane] - Kuznetsova_1990_Ukr.Biokhim.Zh_62_42 |
| Author(s) : Kuznetsova LP , Kugusheva LI , Nikol'skaia EB |
| Ref : Ukr Biokhim Zh , 62 :42 , 1990 |
| Abstract : Kuznetsova_1990_Ukr.Biokhim.Zh_62_42 |
| ESTHER : Kuznetsova_1990_Ukr.Biokhim.Zh_62_42 |
| PubMedSearch : Kuznetsova_1990_Ukr.Biokhim.Zh_62_42 |
| PubMedID: 2087792 |
| Title : Pharmacokinetics of galanthamine (a long-acting anticholinesterase drug) in anaesthetized patients - Westra_1986_Br.J.Anaesth_58_1303 |
| Author(s) : Westra P , van Thiel MJ , Vermeer GA , Soeterbroek AM , Scaf AH , Claessens HA |
| Ref : British Journal of Anaesthesia , 58 :1303 , 1986 |
| Abstract : Westra_1986_Br.J.Anaesth_58_1303 |
| ESTHER : Westra_1986_Br.J.Anaesth_58_1303 |
| PubMedSearch : Westra_1986_Br.J.Anaesth_58_1303 |
| PubMedID: 3778794 |
| Title : [Miniature currents of the endplates of the muscle fibers of the diaphragm of the rat after inhibition of acetylcholinesterase with galanthamine] - Krivoi_1985_Neirofiziologiia_17_607 |
| Author(s) : Krivoi II , Kuleshov VI , Matiushkin DP , Sanotskii VI , Sei TP |
| Ref : Neirofiziologiia , 17 :607 , 1985 |
| Abstract : Krivoi_1985_Neirofiziologiia_17_607 |
| ESTHER : Krivoi_1985_Neirofiziologiia_17_607 |
| PubMedSearch : Krivoi_1985_Neirofiziologiia_17_607 |
| PubMedID: 2999623 |
| Title : Homer's moly identified as Galanthus nivalis L.: physiologic antidote to stramonium poisoning - Plaitakis_1983_Clin.Neuropharmacol_6_1 |
| Author(s) : Plaitakis A , Duvoisin RC |
| Ref : Clinical Neuropharmacology , 6 :1 , 1983 |
| Abstract : Plaitakis_1983_Clin.Neuropharmacol_6_1 |
| ESTHER : Plaitakis_1983_Clin.Neuropharmacol_6_1 |
| PubMedSearch : Plaitakis_1983_Clin.Neuropharmacol_6_1 |
| PubMedID: 6342763 |
| Title : Reversal of central anticholinergic syndrome by galanthamine - Baraka_1977_Jama_238_2293 |
| Author(s) : Baraka A , Harik S |
| Ref : Jama , 238 :2293 , 1977 |
| Abstract : Baraka_1977_Jama_238_2293 |
| ESTHER : Baraka_1977_Jama_238_2293 |
| PubMedSearch : Baraka_1977_Jama_238_2293 |
| PubMedID: 578850 |
| Title : [Characteristics of galanthamine as a reversible inhibitor of cholinesterase] - |
| Author(s) : Vasilenko ET , Tonkopii VD |
| Ref : Biokhimiia , 39 :701 , 1974 |
| PubMedID: 4441566 |
| Title : Studies on the synthesis of heterocyclic compounds. CCCXV. Modified total synthesis of (plus or minus)-galanthamine through phenol oxidation - |
| Author(s) : Kametani T , Yamaki K , Yagi H , Fukumoto K |
| Ref : J Chem Soc Perkin 1 , 18 :2602 , 1969 |
| PubMedID: 5391300 |
| Title : [Influence exerted by galanthamine on the acetylcholinesterase activity of various regions of the brain] - |
| Author(s) : Nesterenko LN |
| Ref : Farmakologiia i Toksikologiia , 28 :413 , 1965 |
| PubMedID: 5868403 |
| Title : On the pharmacology of the new alkaloid galantamine - |
| Author(s) : Mashkovsky MD , Kruglikova-Lvova RP |
| Ref : Farmakologia Toxicologia (Moskow) , 14 :27 , 1951 |
| PubMedID: |